HC - IBD label transfer
Last compiled on 17 November, 2022
We want to compare the clusters we obtained with the two datasets:
Libraries
library(matchSCore2)
library(nnet)
library(Matrix)
library(Seurat)
library(ggplot2)Data preparation
subsets <- c('epi', 'myeloids', 'plasmas', 'stroma', 'tcells')
for(subset in subsets){
ref <- readRDS(paste0("~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/01_Samples_by_Health/HC/annotated/", subset, ".RDS"))
ref@active.ident <- factor(ref$annotation)
output.seu <- FindAllMarkers(ref, only.pos = T)
gene_cl.ref <- cut_markers(levels(output.seu$cluster),output.seu,ntop=100)
saveRDS(gene_cl.ref, file = paste0("~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/01_Samples_by_Health/HC/annotated/genes/",subset,"_gene_cl.ref.RDS"))
}
for(subset in subsets){
ref <- readRDS(paste0("~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/01_Samples_by_Health/IBD/annotated/", subset, ".RDS"))
ref@active.ident <- factor(ref$annotation)
output.seu <- FindAllMarkers(ref, only.pos = T)
gene_cl.ref <- cut_markers(levels(output.seu$cluster), output.seu, ntop=100)
saveRDS(gene_cl.ref, file = paste0("~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/01_Samples_by_Health/IBD/annotated/genes/",subset,"_gene_cl.ref.RDS"))
}Jaccard indexes
for(subset in subsets){
ibd <- readRDS(file = paste0("~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/01_Samples_by_Health/IBD/annotated/genes/",
subset,
"_gene_cl.ref.RDS"))
hc <- readRDS(file = paste0("~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/01_Samples_by_Health/HC/annotated/genes/",
subset,
"_gene_cl.ref.RDS"))
ms <- matchSCore2(gene_cl.ref = hc,
gene_cl.obs = ibd,
ylab = "IBD",
xlab = "HC")
ms$ggplot$layers[[2]]$aes_params$size <- 3
j <- ms$ggplot +
theme(axis.text.x = element_text(angle=90,
vjust = 0.5, size = 9),
axis.text.y = element_text(size = 9))
cat("## ", subset, "\n"); print(j); cat("\n\n")
}epi
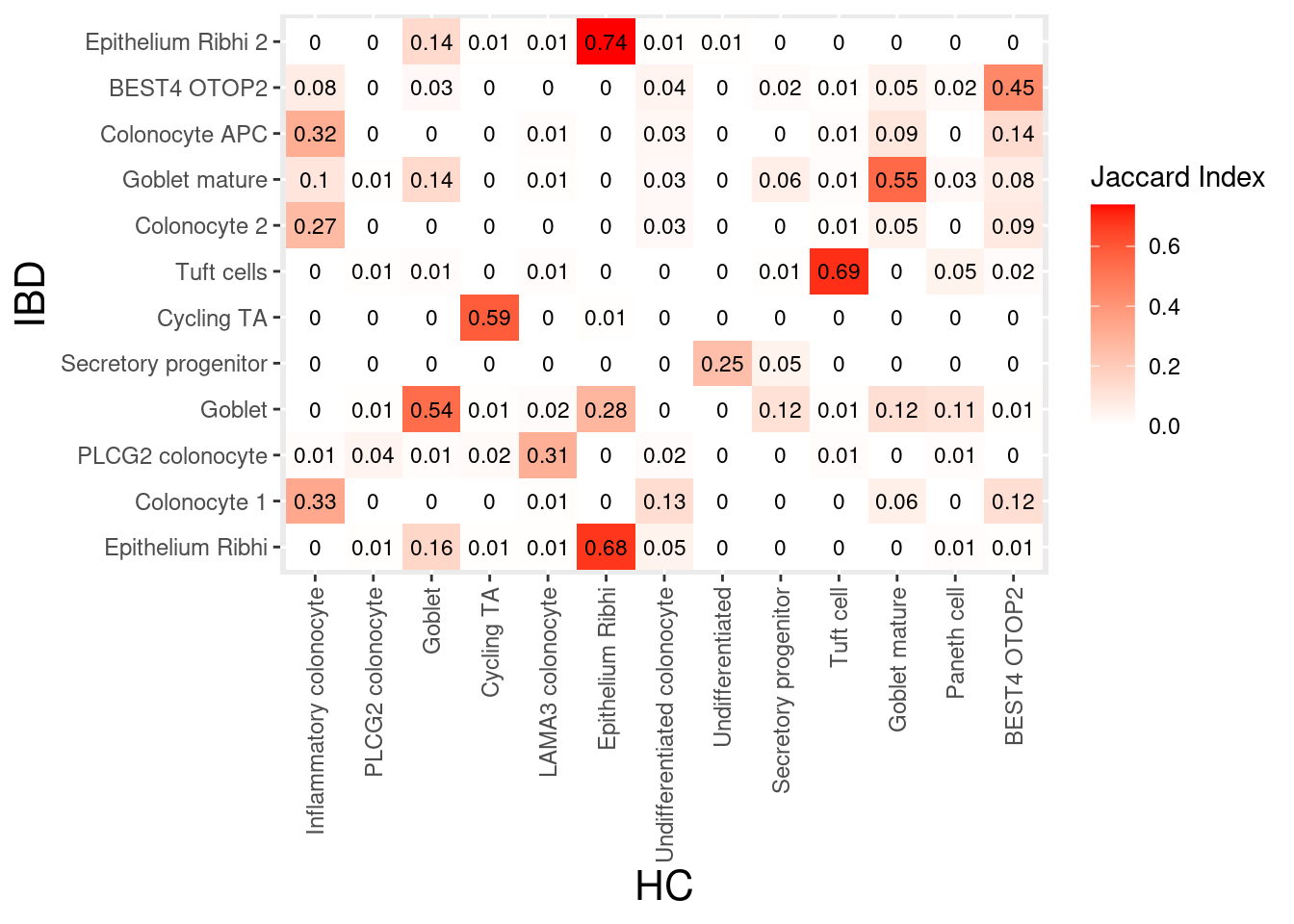
myeloids
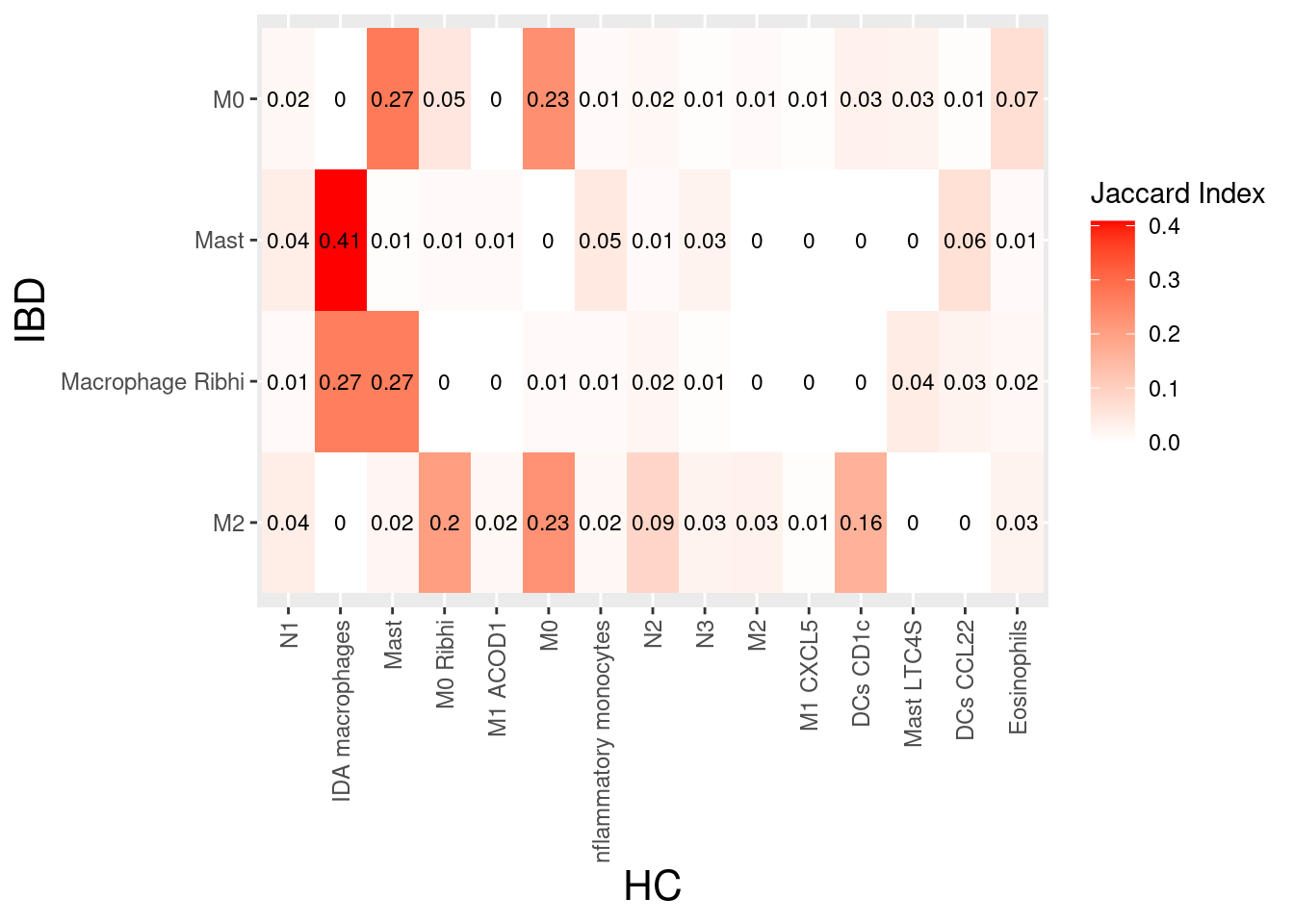
plasmas
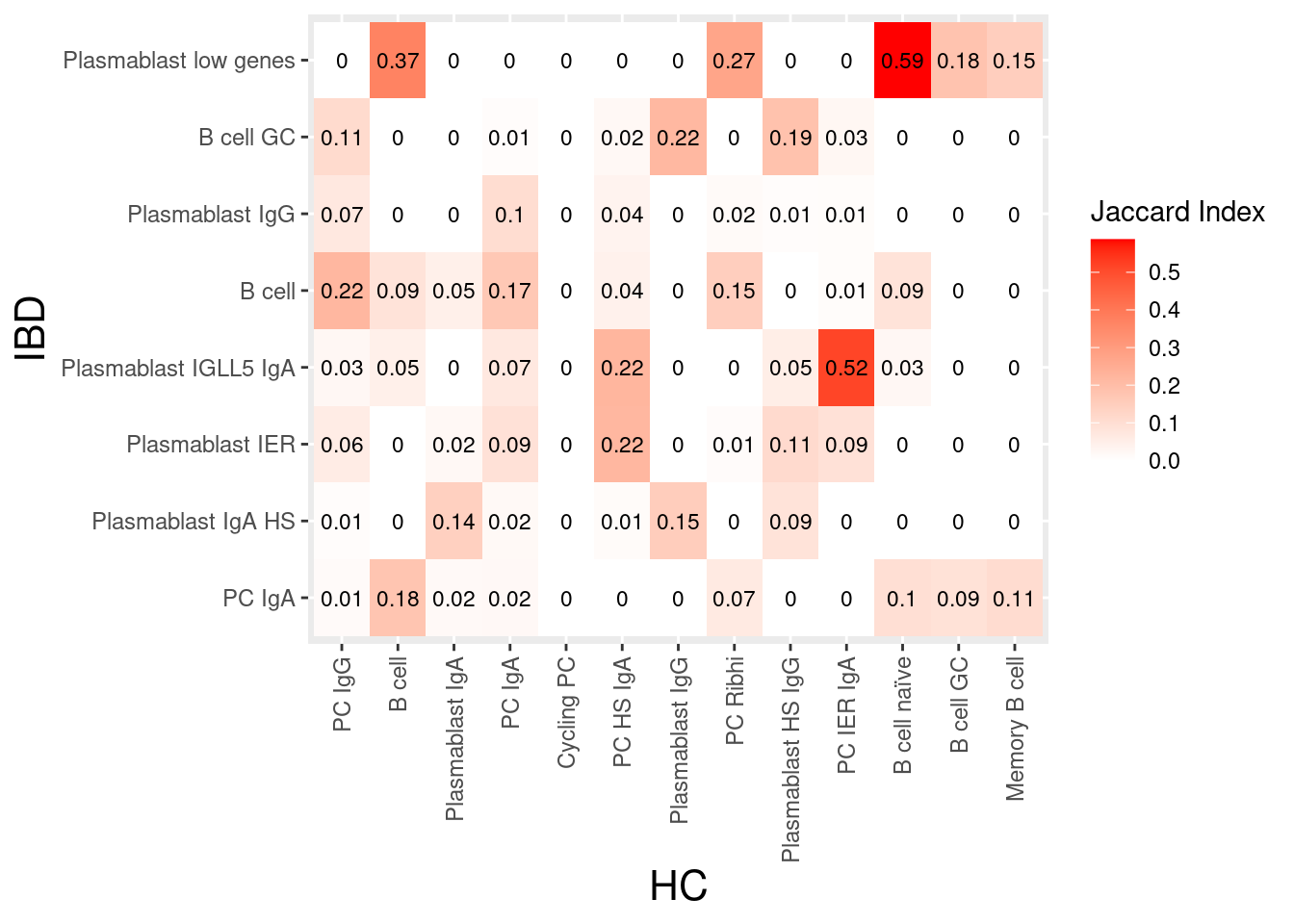
stroma
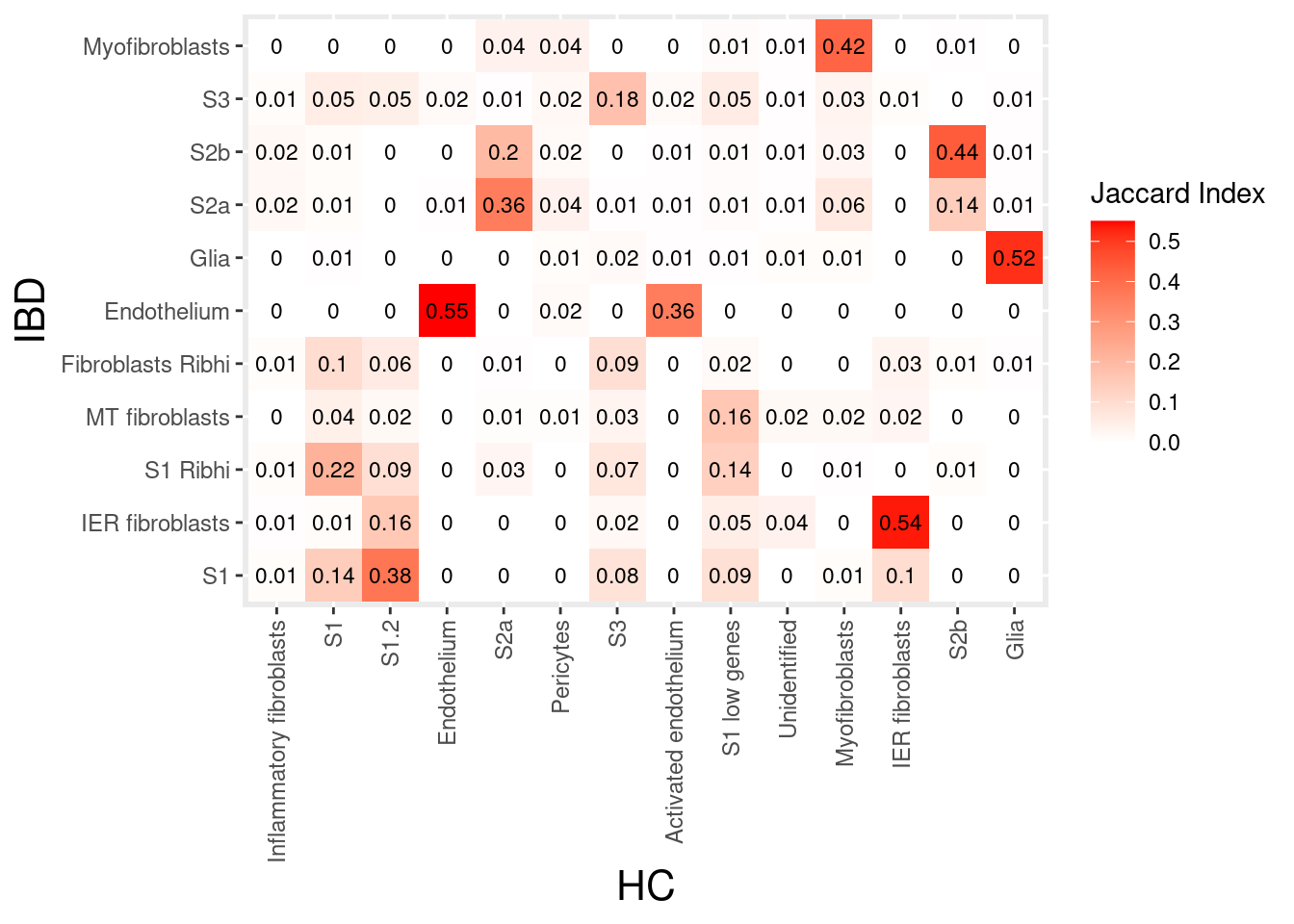
tcells
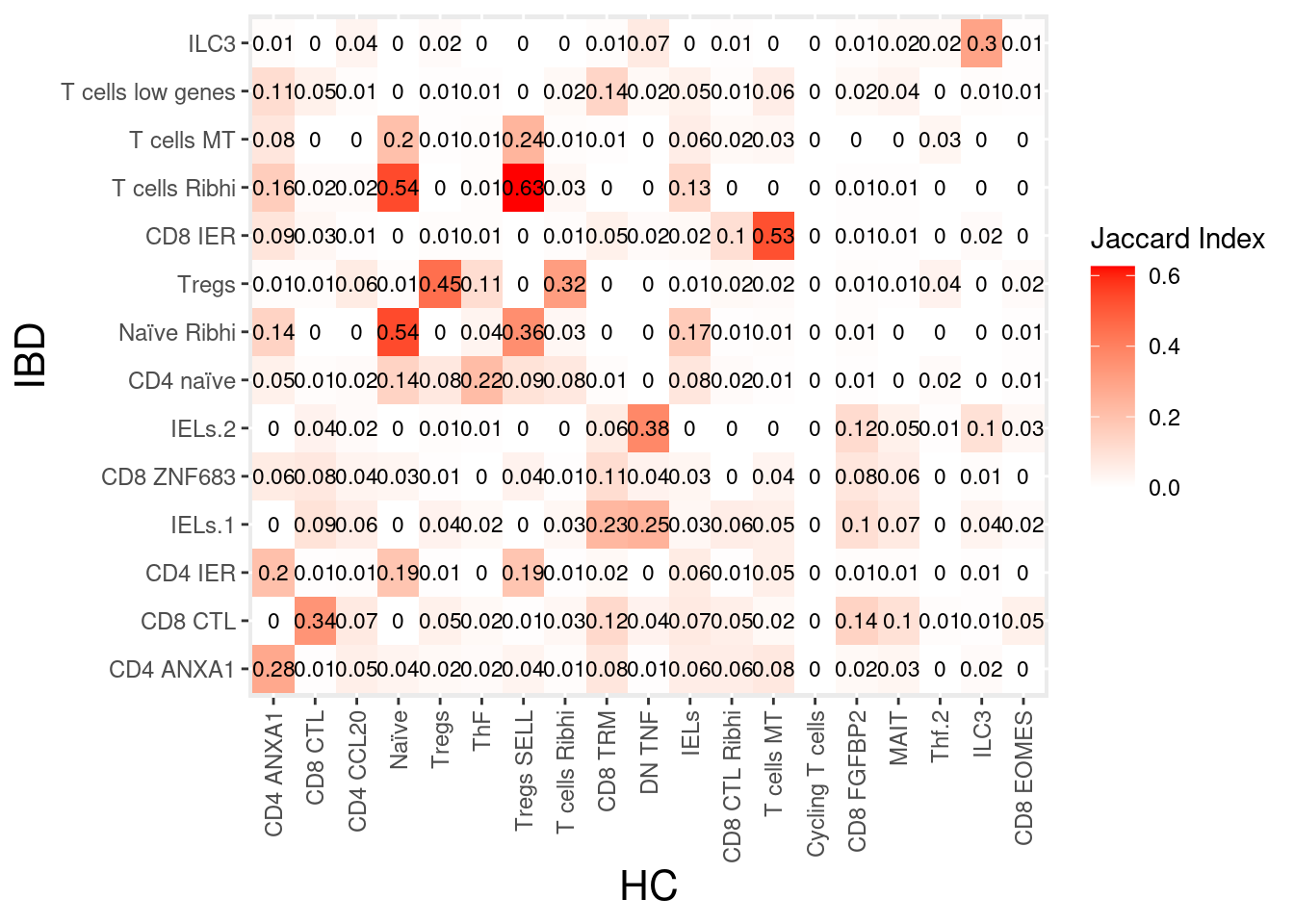
```