Split the data by subset
Last compiled on 17 November, 2022
We considered five major cell subsets: (1) myeloid, (2) T, (3) B and plasma, (4) stromal, and (5) epithelial cells. We classified cells into these subsets according to their gene expression.
Load libraries
library(Seurat)
library(plyr)
library(dplyr)
library(ggplot2)
library(DropletUtils)
library(celda)
library(SingleCellExperiment)
library(scater)
library(scran)
library(scDblFinder)
library(viridis)
library(MASS)
library(patchwork)
library(readr)Load extra sources
get_density <- function(x, y, ...) { # function from https://slowkow.com/notes/ggplot2-color-by-density/
dens <- MASS::kde2d(x, y, ...)
ix <- findInterval(x, dens$x)
iy <- findInterval(y, dens$y)
ii <- cbind(ix, iy)
return(dens$z[ii])
}
fancy_scientific <- function(l) { # function from https://stackoverflow.com/a/24241954
# turn in to character string in scientific notation
l <- format(l, scientific = TRUE)
# quote the part before the exponent to keep all the digits
l <- gsub("^(.*)e", "'\\1'e", l)
# turn the 'e+' into plotmath format
l <- gsub("e", "%*%10^", l)
# return this as an expression
parse(text=l)
}
setwd("~/000_GitHub/ibd-bcn_single_cell")
source('source/functions_scrnaseq.R')
source('source/colors.R')Load the data
setwd('~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/')
seudataf <- readRDS(file = 'filtered_data_together.RDS')Dimension reduction
We cluster the samples using louvain classification and look for the markers.
setwd('~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/')
seudataf <- seurat_to_pca(seudataf)
a <- ElbowPlot(seudataf, ndims = 50) +
geom_vline(xintercept = 23, colour="#BB0000", linetype = 2)+
labs(title = paste0('Elbowplot'))+ theme_classic()
seudataf <- FindNeighbors(seudataf, dims = 1:23)
seudataf <- RunUMAP(seudataf, dims = 1:23)
b <- DimPlot(seudataf, group.by = 'sample_name', pt.size = 0.1) +
labs(title = '23 PCS') +
theme_classic() +
theme(legend.position = 'bottom')
(a+b) / guide_area() +
plot_layout(heights = c(0.7,0.3),guides = 'collect')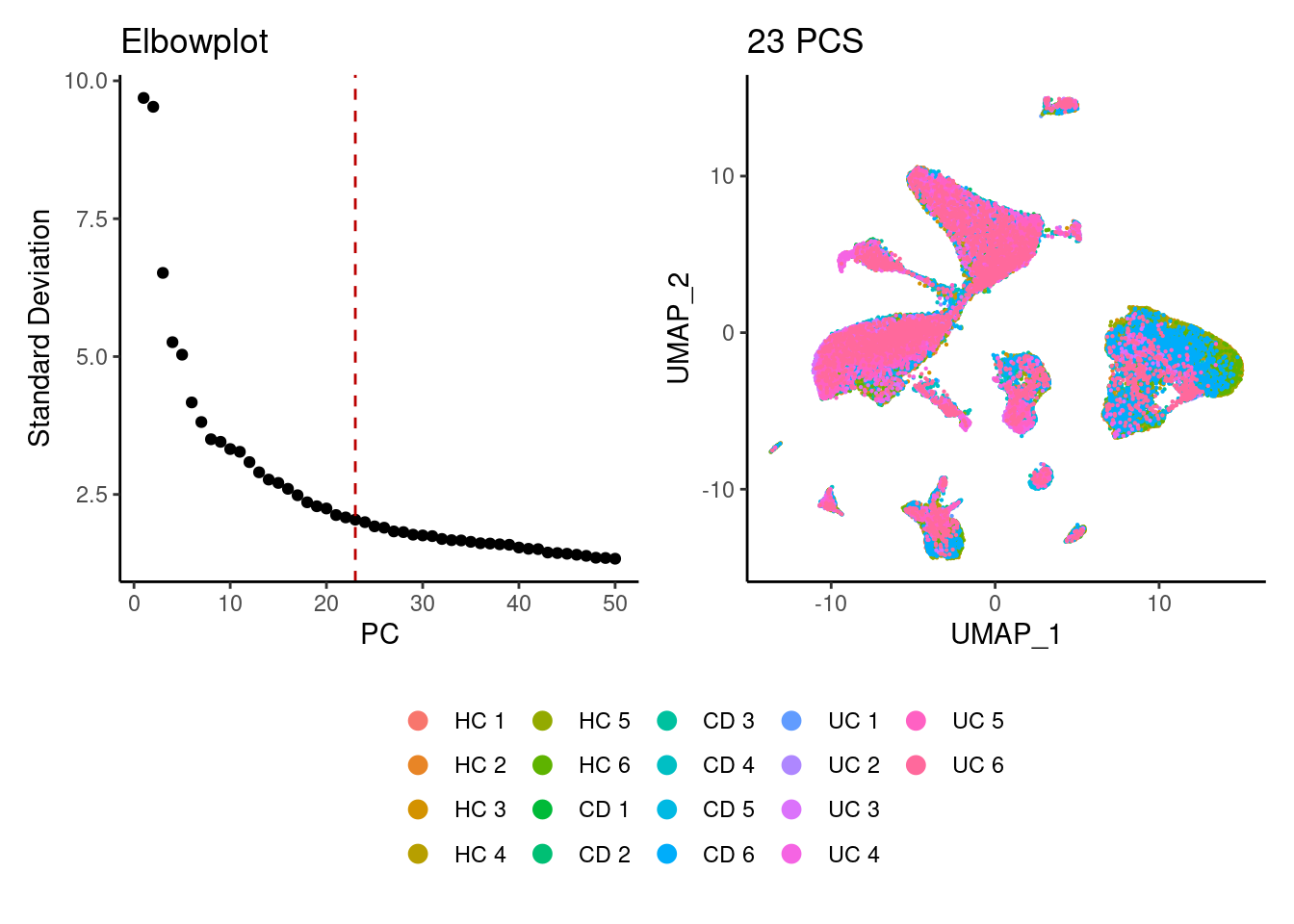
dir.create('ALL')
dir.create('ALL/filter_65')
seudataf <- resolutions(seudataf, resolutions = c(0.1, 0.3, 0.5),
workingdir = 'ALL/filter_65/',
title = 'ALL')
saveRDS(seudataf,'ALL/filter_65/ALL.RDS')Resolutions plot
a <- DimPlot(seudataf, group.by = 'RNA_snn_res.0.1', label = T) + theme_classic() + NoLegend()
b <- DimPlot(seudataf, group.by = 'RNA_snn_res.0.3', label = T) + theme_classic() + NoLegend()
c <- DimPlot(seudataf, group.by = 'RNA_snn_res.0.5', label = T) + theme_classic() + NoLegend()
(a + b) / (c + plot_spacer() )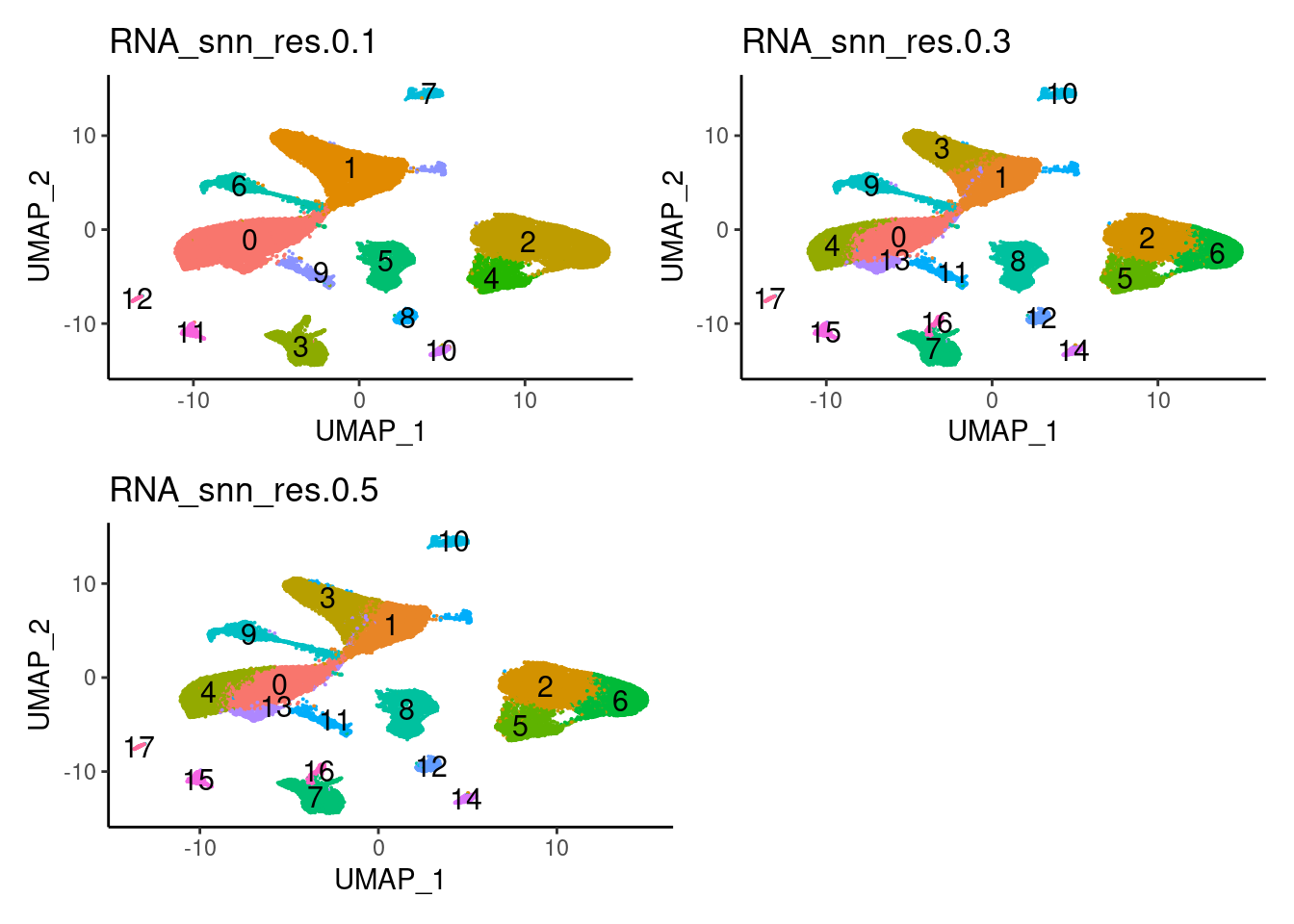
Top5 markers
marker_genes <- read_delim("ALL/filter_65/ALL_markers_resolution_0.1.csv",
delim = ";", escape_double = FALSE,
locale = locale(decimal_mark = ",", grouping_mark = "."),
trim_ws = TRUE)## Rows: 9064 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.top5 <- marker_genes %>%
dplyr::group_by(cluster)%>%
dplyr::slice(1:5)
top5_g <- unique(top5$gene)
DotPlot(seudataf, features = top5_g, group.by = 'RNA_snn_res.0.1' ) + theme_classic() + theme(axis.text.x = element_text(angle=90), axis.title = element_blank()) + NoLegend() + labs(title = 'Resolution 0.1')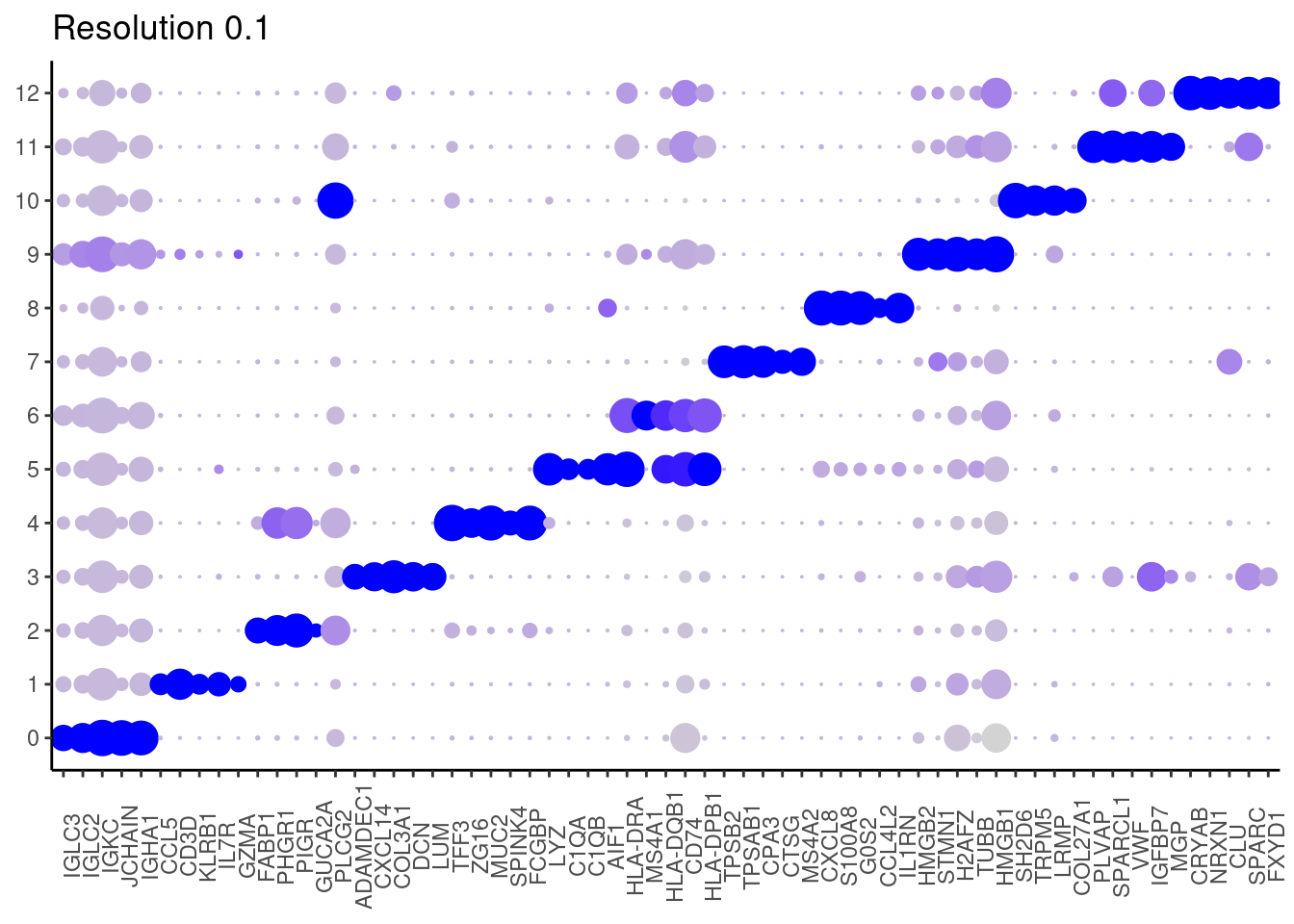
Bibliography genes
subset_genes <- data.frame(
stringsAsFactors = FALSE,
gene = c("BANK1","CD19","CD79A","DERL3", # b and plasma
"MS4A1","MZB1",
"AQP8","BEST4", "EPCAM","MUC2", # epi
"OLFM4","TRPM5", "ZG16",
"AIF1","C1QA","C1QB","CD14", # myeloids
"CMTM2","FCGR3B","LYZ","MS4A2",
"TPSAB1","TPSB2", 'CPA3',
'CD14', 'CD163', 'CSF1R', 'CD68',
"ACTA2","ADAMDEC1","CHI3L1", # stroma
"COL3A1","NRXN1","PLVAP","SOX6",
"VWF",
'PDGFRA', 'PDGFRB', 'PTGDS', 'LUM', 'MMP2',
"CD3D","CD3E","CD3G", # tcells
"CD8A","FOXP3","GZMA","GZMB",
"TRAC", "NKG7","TRBC1",
'KLRB1'),
subset = c("B and plasmacells","B and plasmacells",
"B and plasmacells",
"B and plasmacells","B and plasmacells",
"B and plasmacells",
"Epithelium",
"Epithelium","Epithelium",
"Epithelium","Epithelium",
"Epithelium","Epithelium",
"Myeloid cells","Myeloid cells","Myeloid cells",
"Myeloid cells","Myeloid cells","Myeloid cells",
"Myeloid cells","Myeloid cells","Myeloid cells",
"Myeloid cells","Myeloid cells","Myeloid cells",
"Myeloid cells","Myeloid cells","Myeloid cells",
"Stromal cells","Stromal cells",
"Stromal cells","Stromal cells","Stromal cells",
"Stromal cells","Stromal cells","Stromal cells",
"Stromal cells","Stromal cells","Stromal cells",
"Stromal cells","Stromal cells",
"T cells","T cells",
"T cells","T cells","T cells",
"T cells","T cells","T cells",
"T cells","T cells", 'NK or NKT cells')
)
for(subset in unique(subset_genes$subset)){
cat("### ", subset, "{.tabset} \n\n")
genes <- subset_genes$gene[subset_genes$subset == subset]
for (gene in genes) {
j <- FeaturePlot(seudataf, features = gene, order=T) + theme_classic()
cat("#### ", gene, "\n"); print(j); cat("\n\n")
}
}B and plasmacells
BANK1
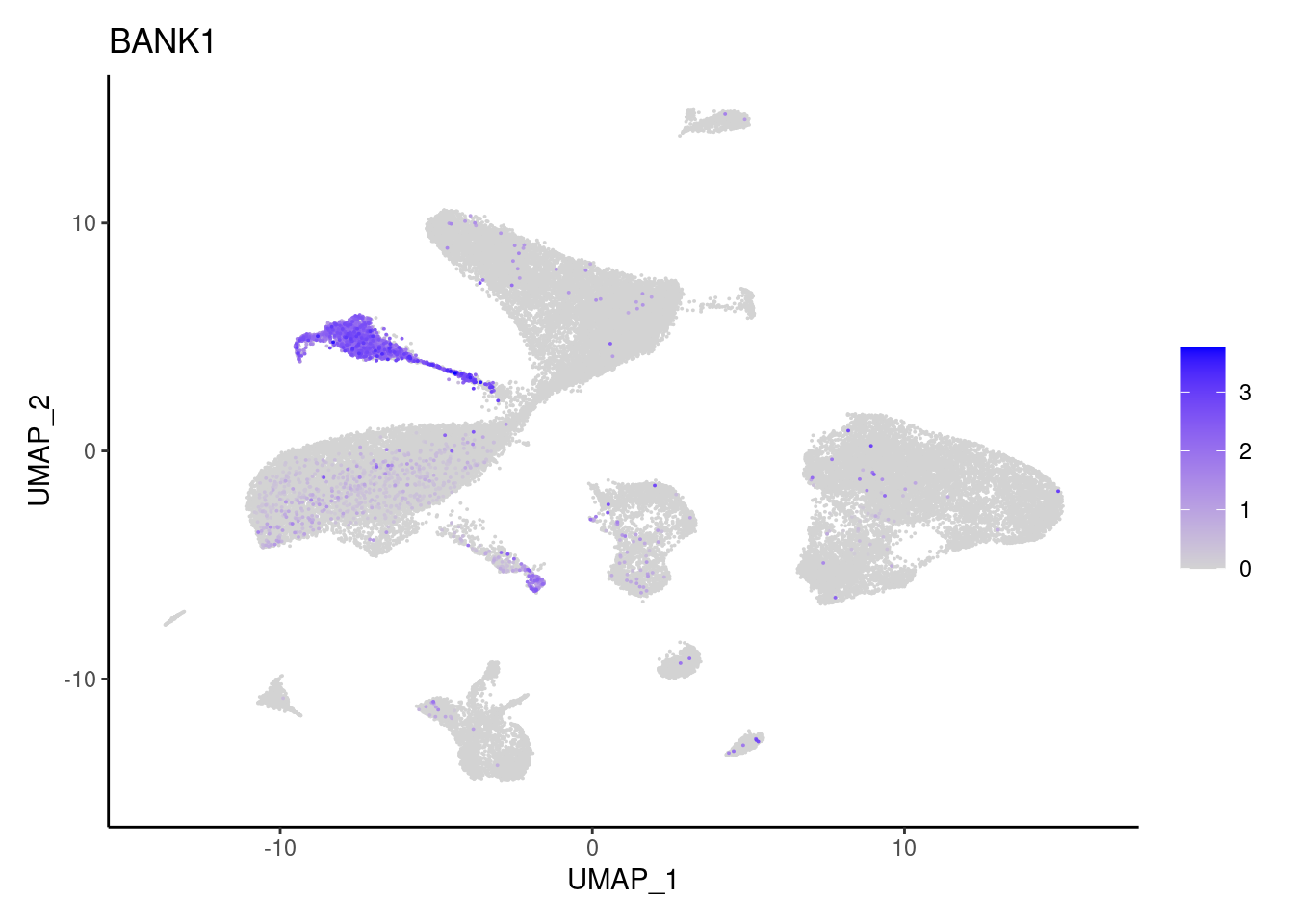
CD19
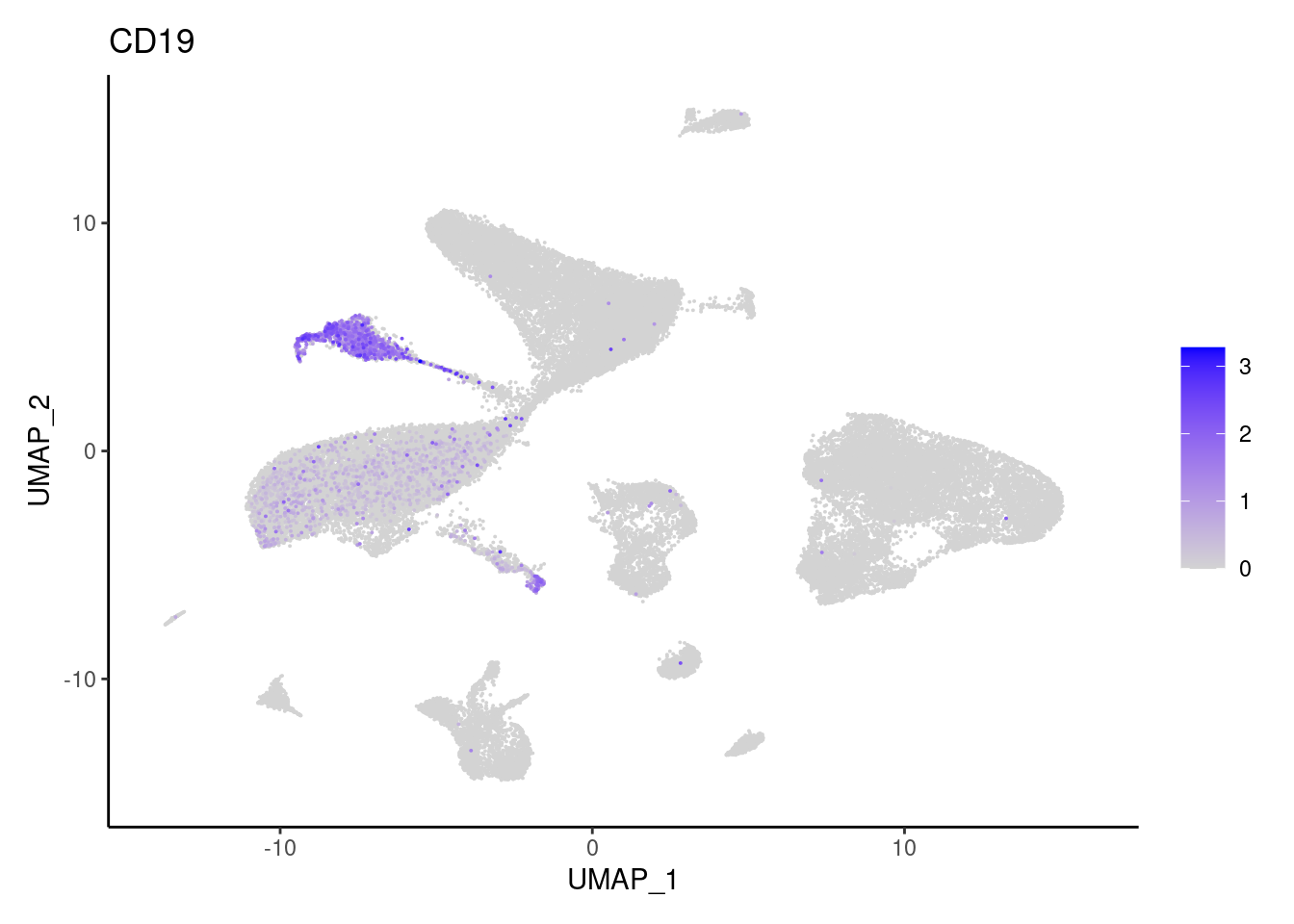
CD79A
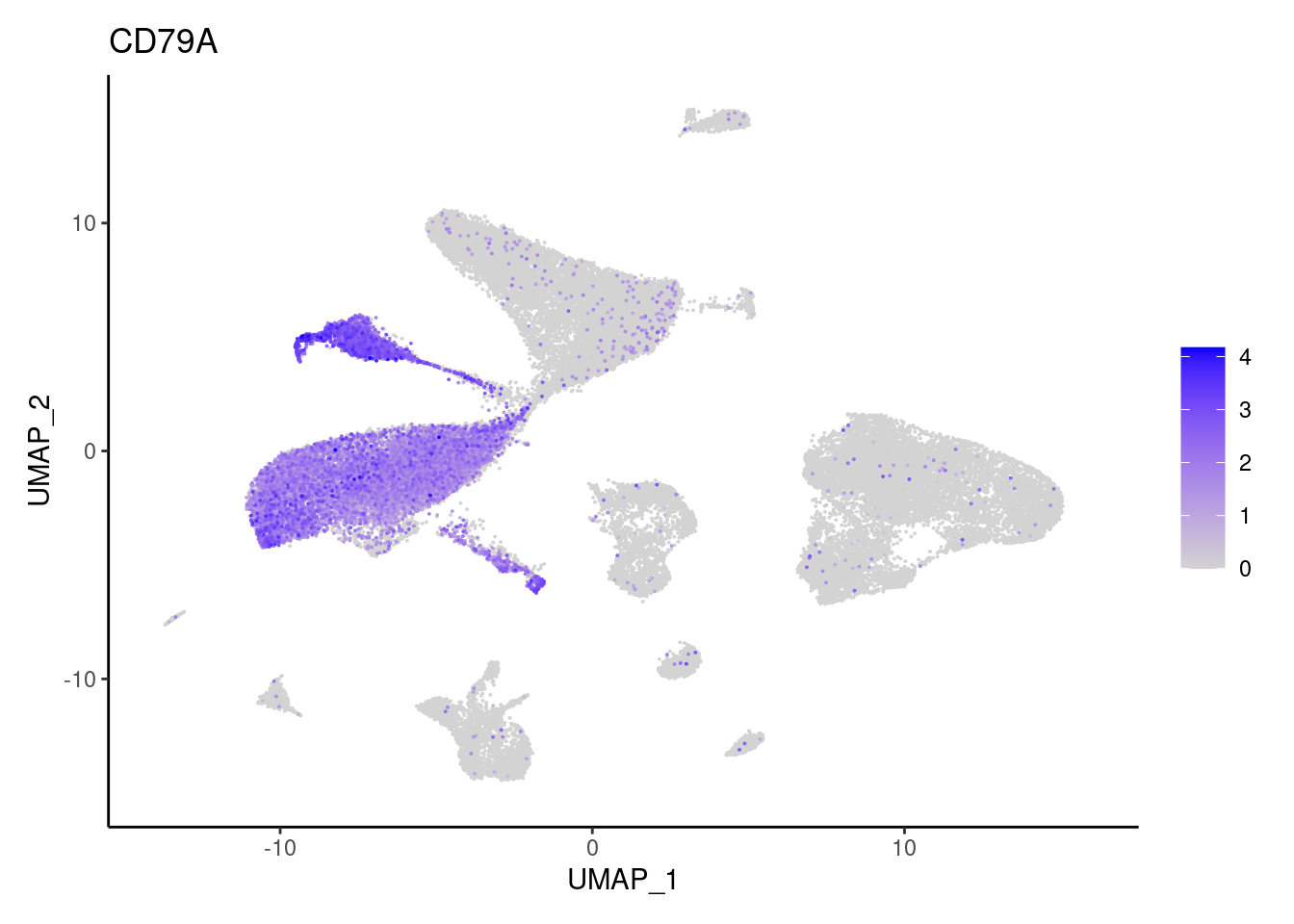
DERL3
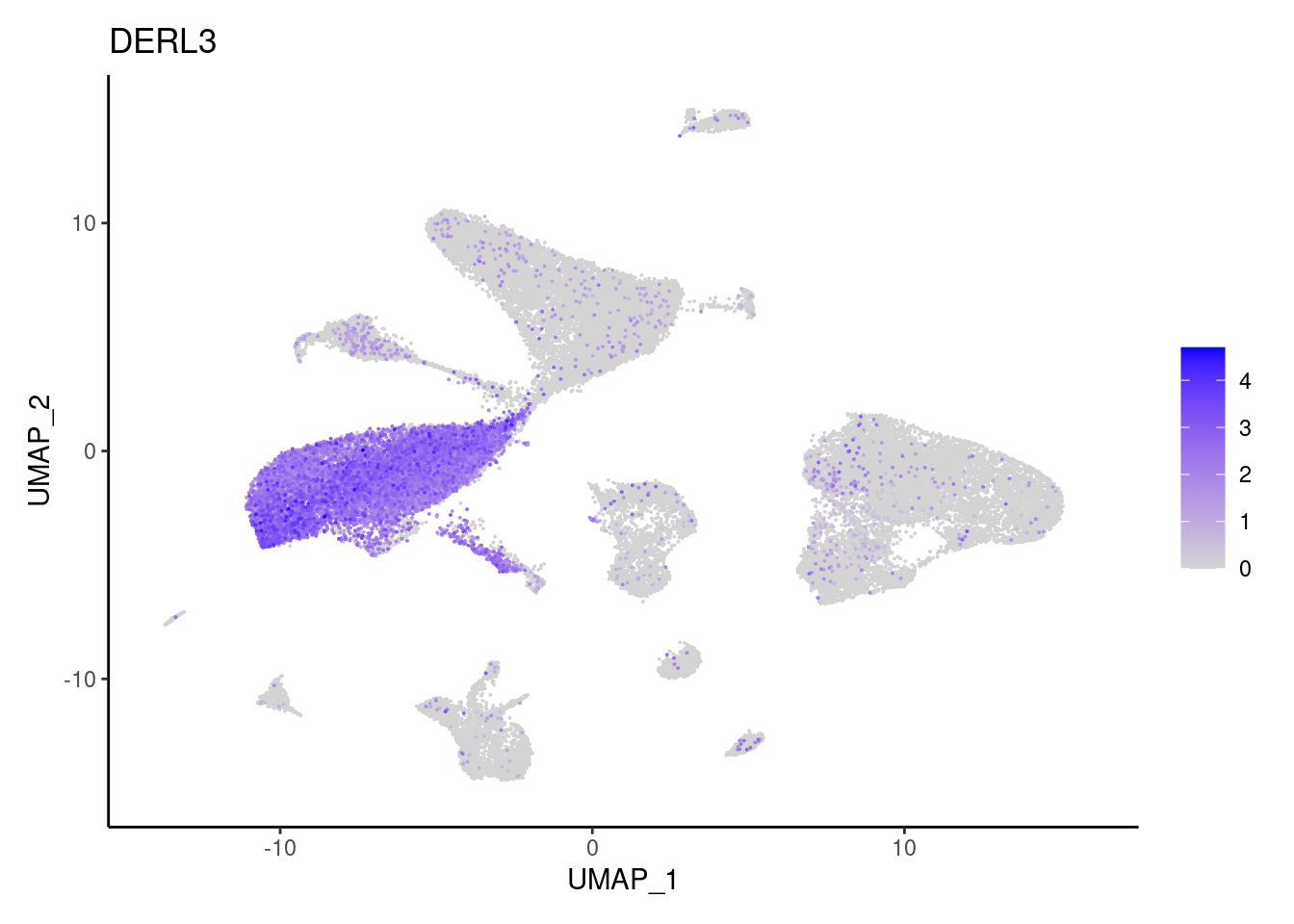
MS4A1
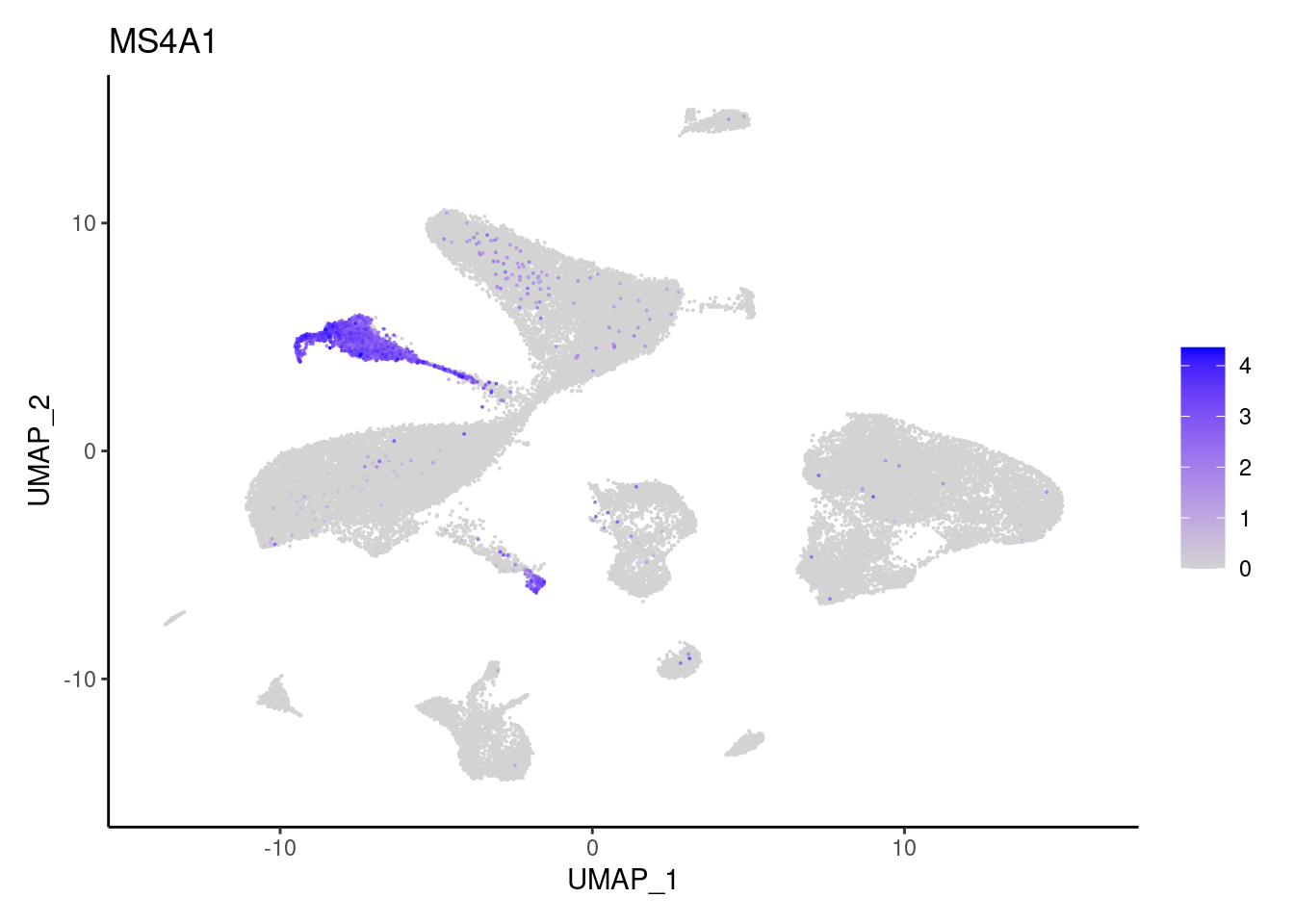
MZB1
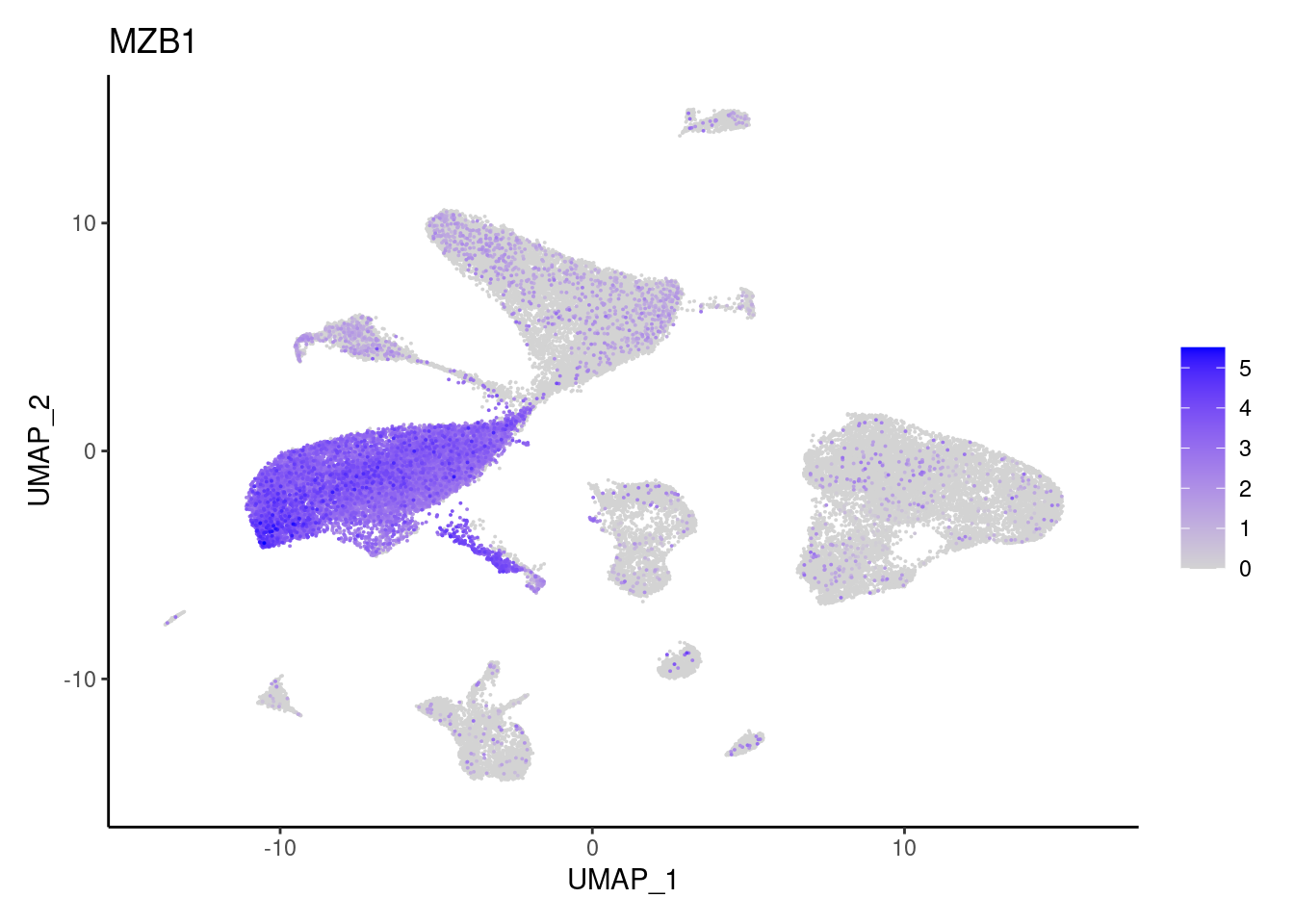
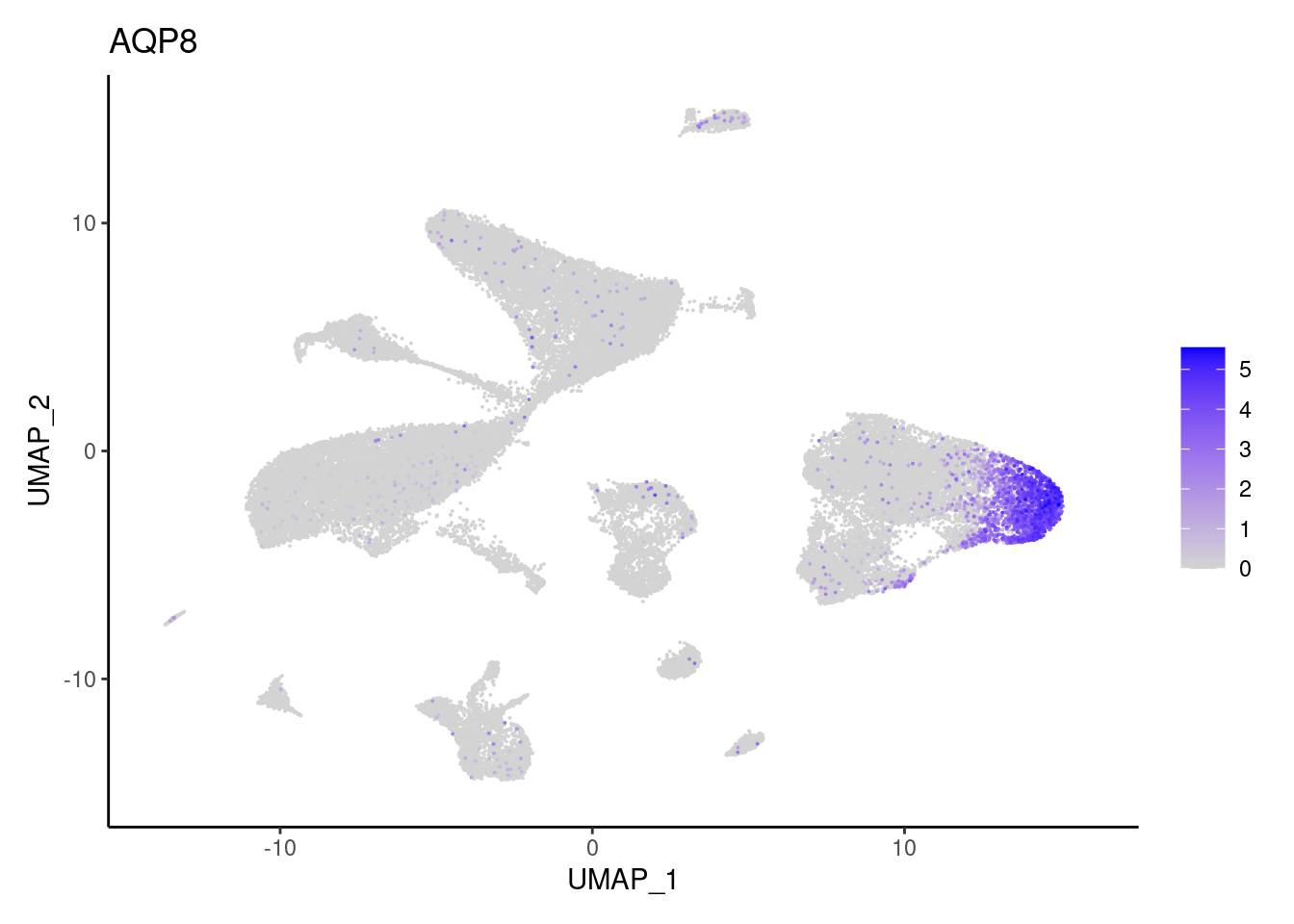
Epithelium
AQP8
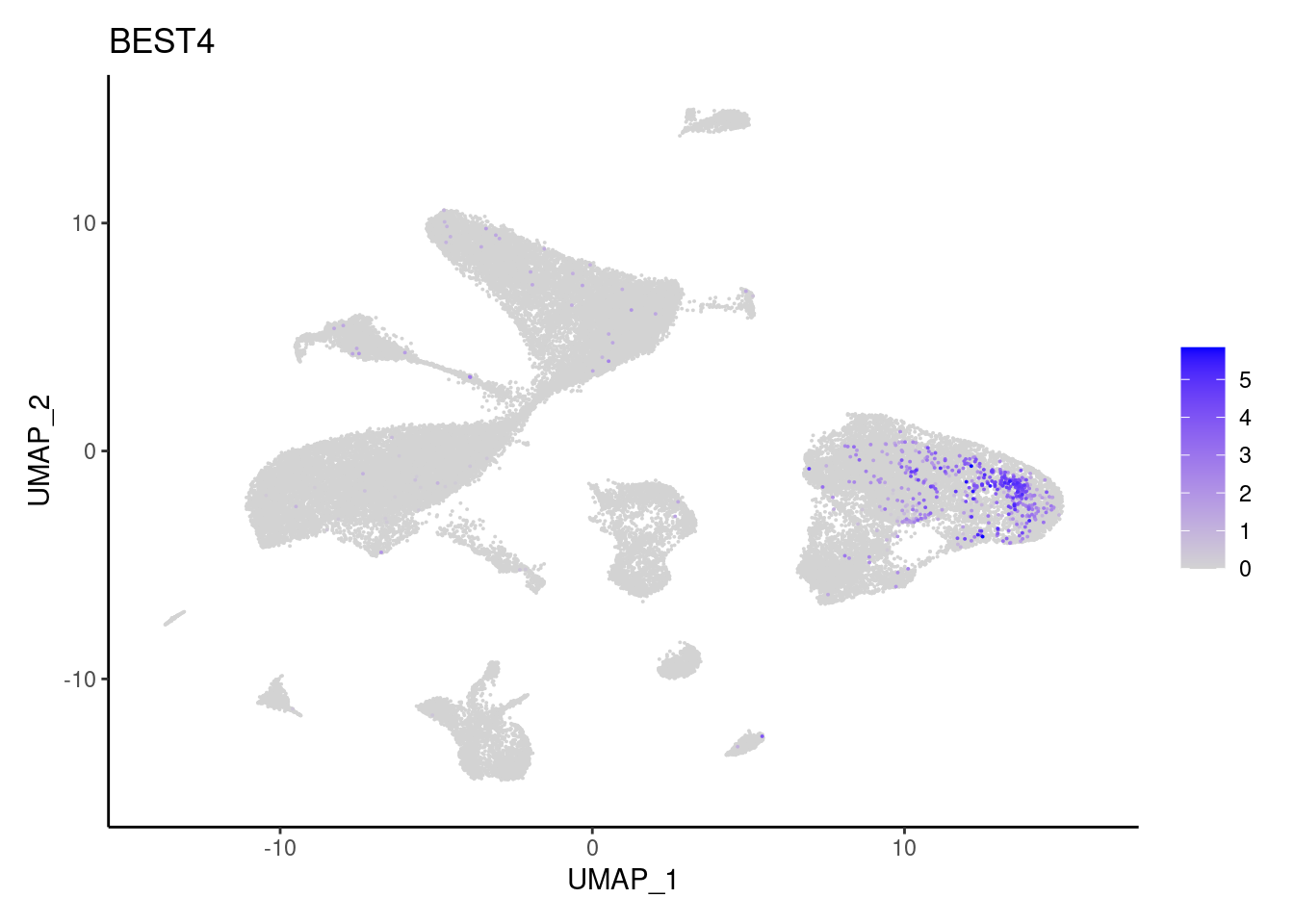
BEST4
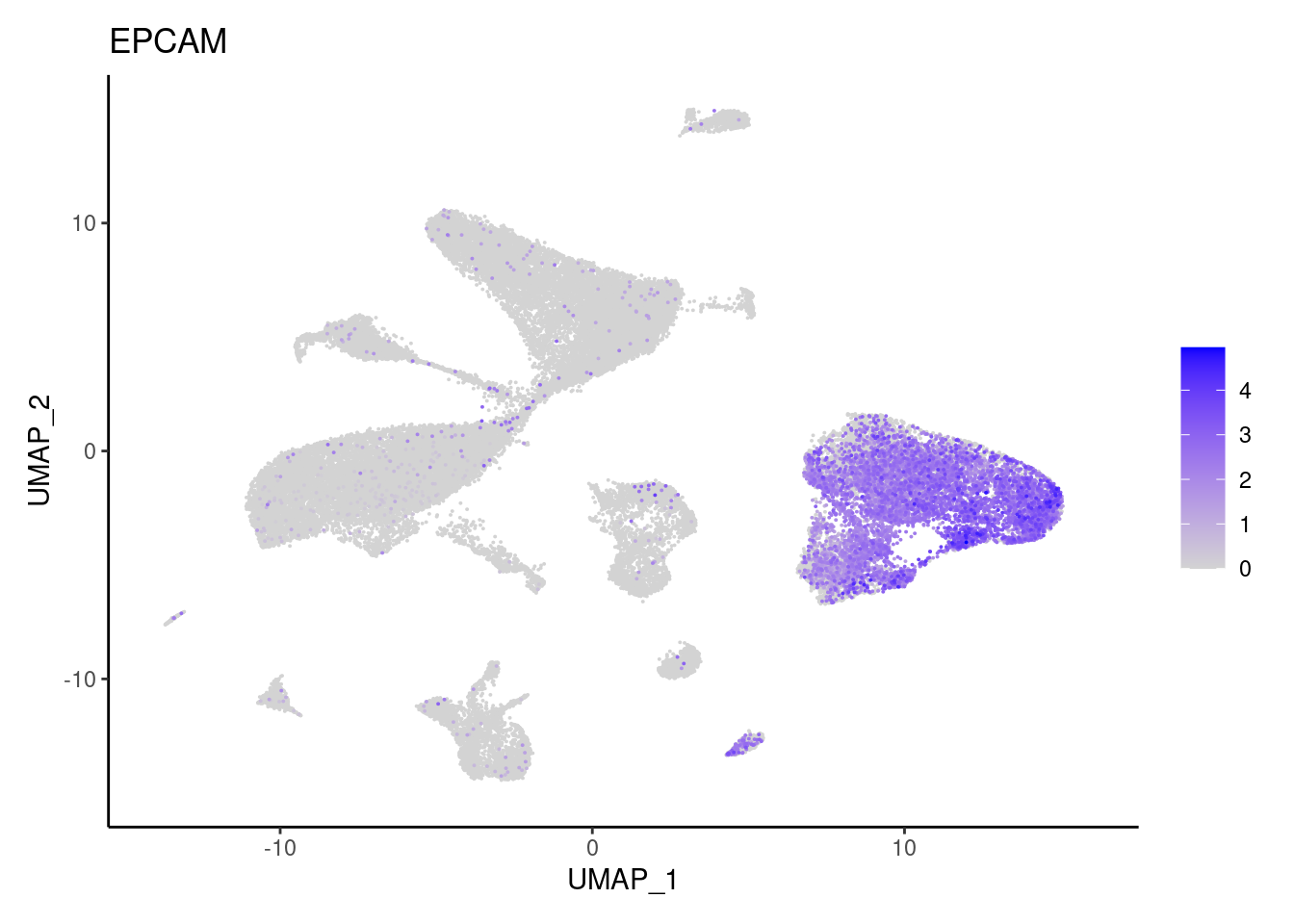
EPCAM
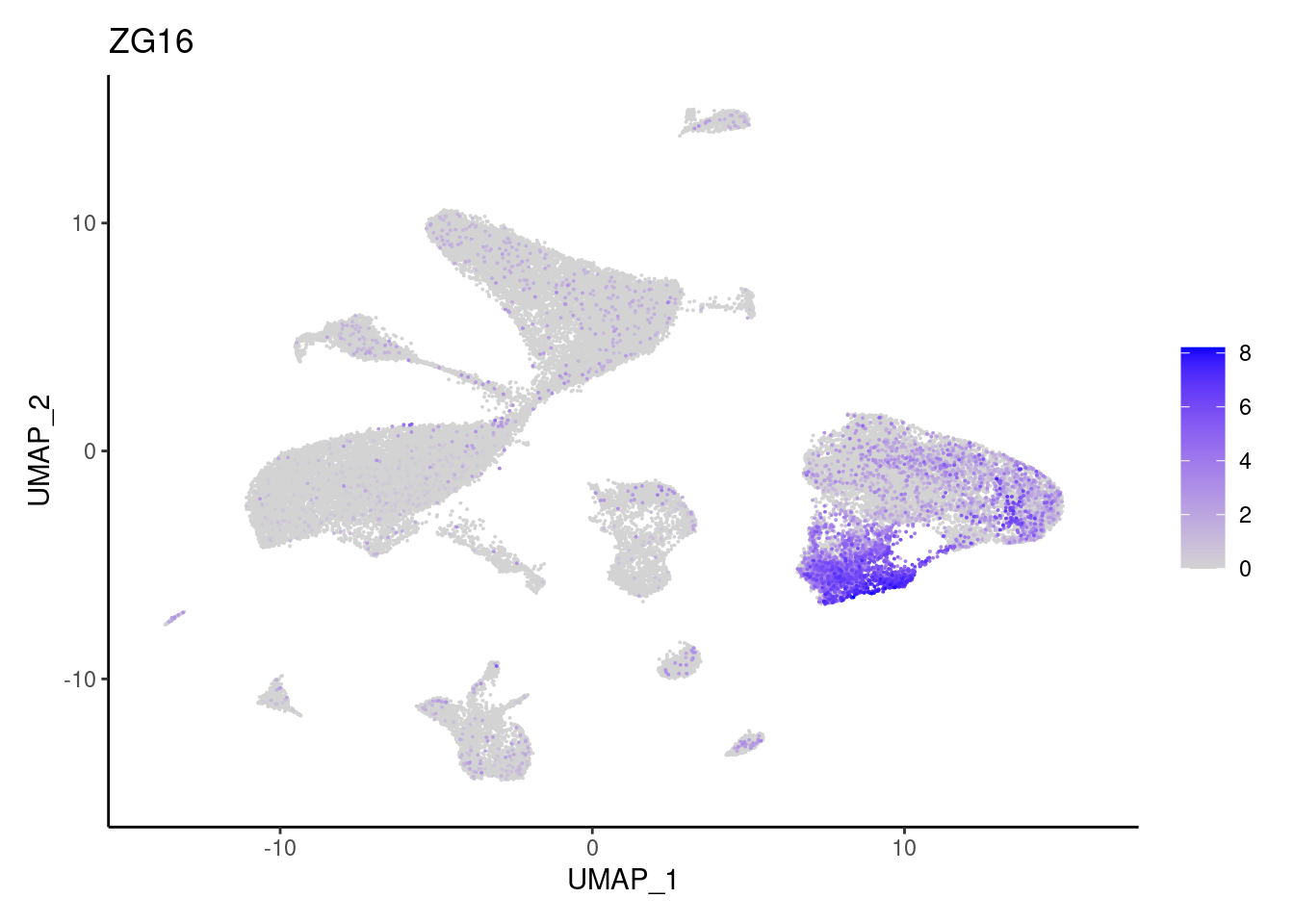
MUC2
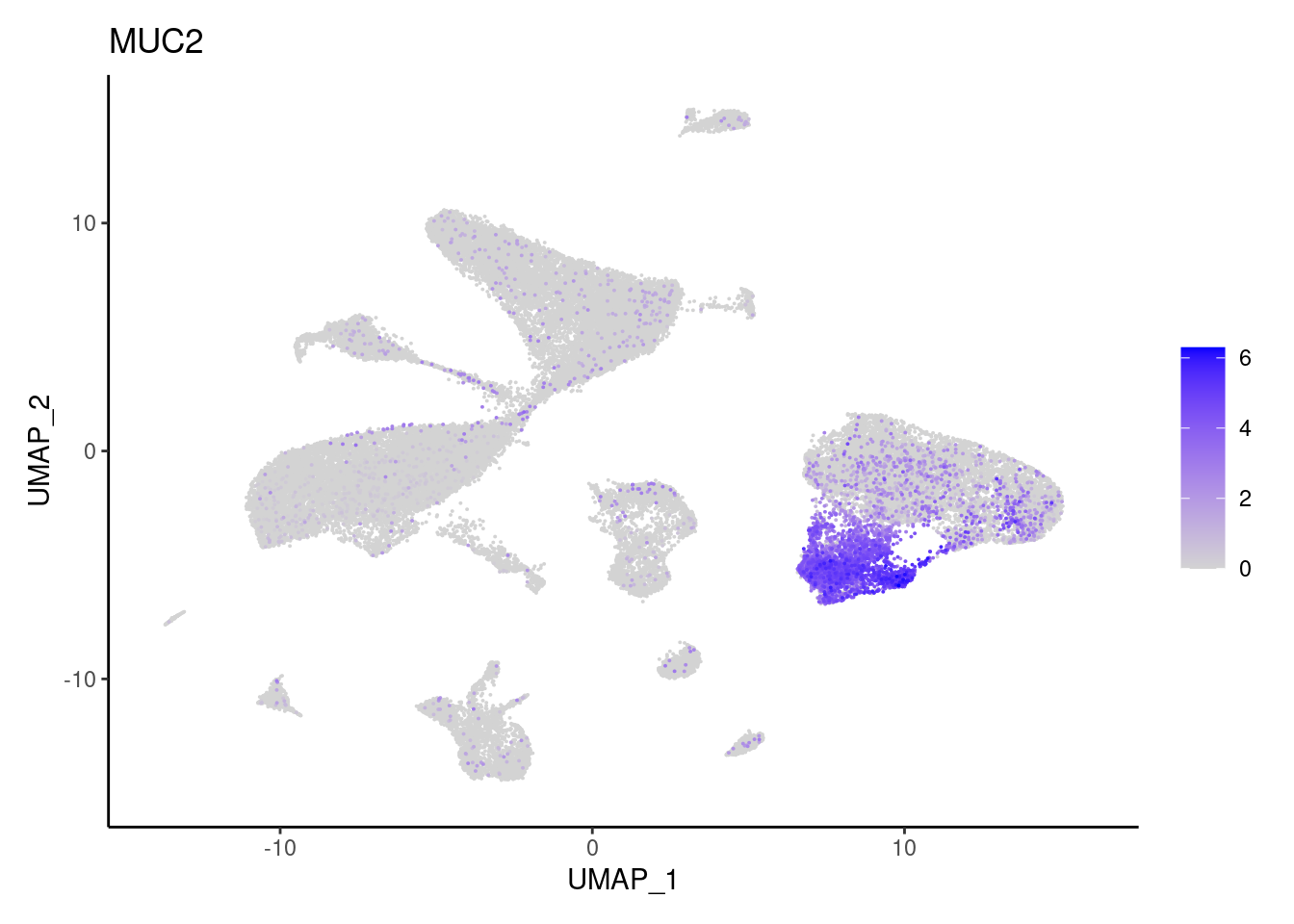
OLFM4
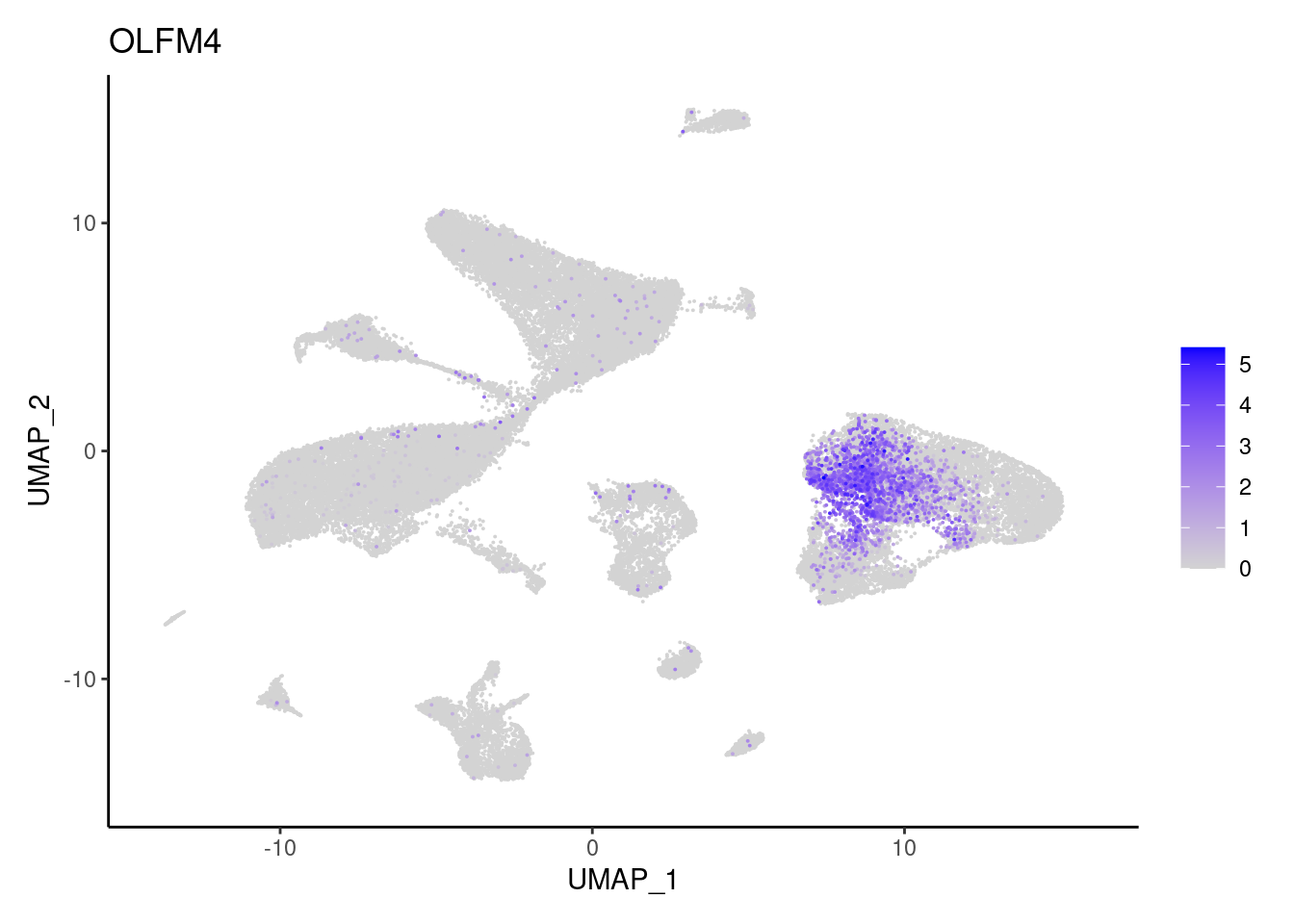
TRPM5
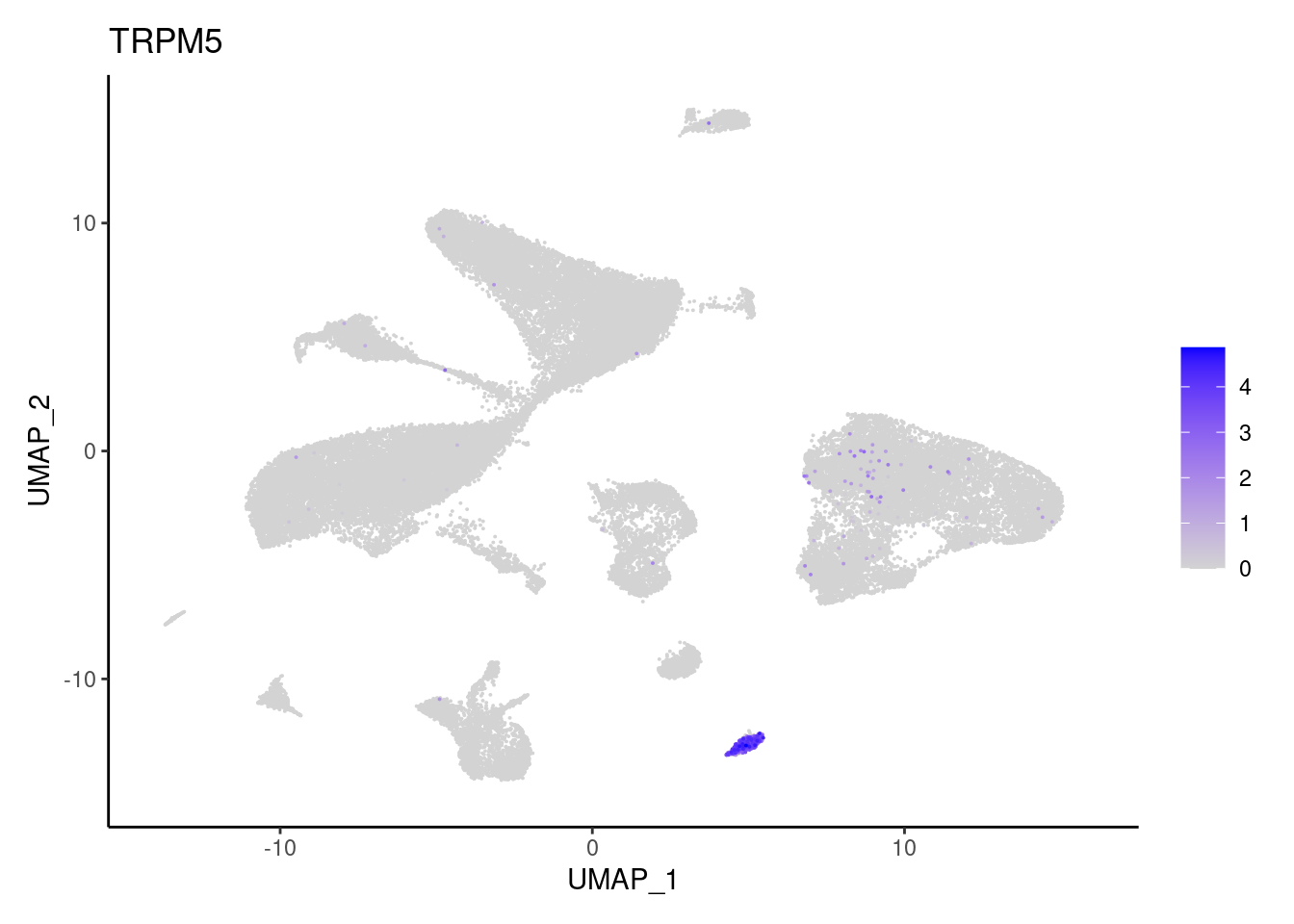
ZG16
Myeloid cells
AIF1
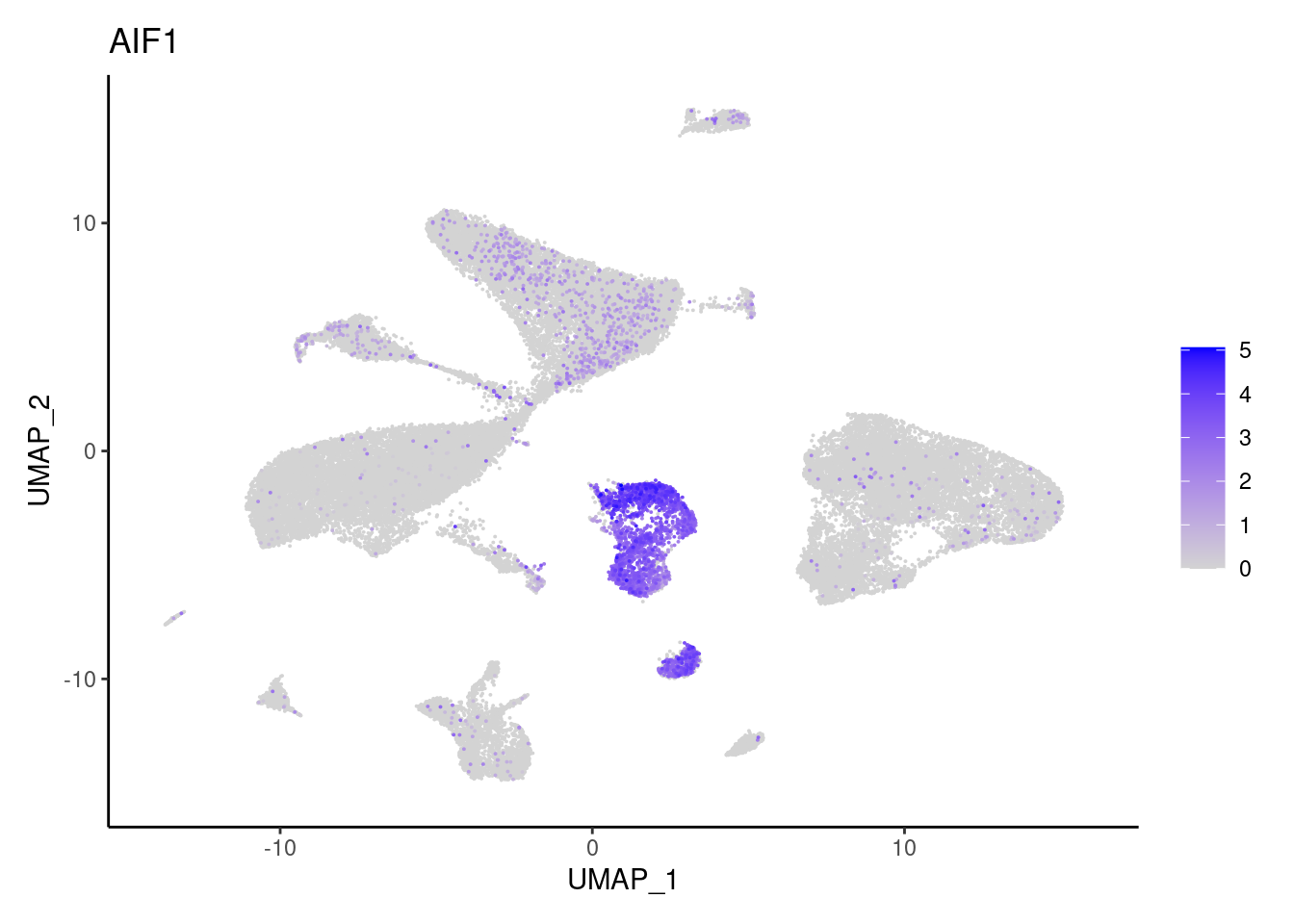
C1QA
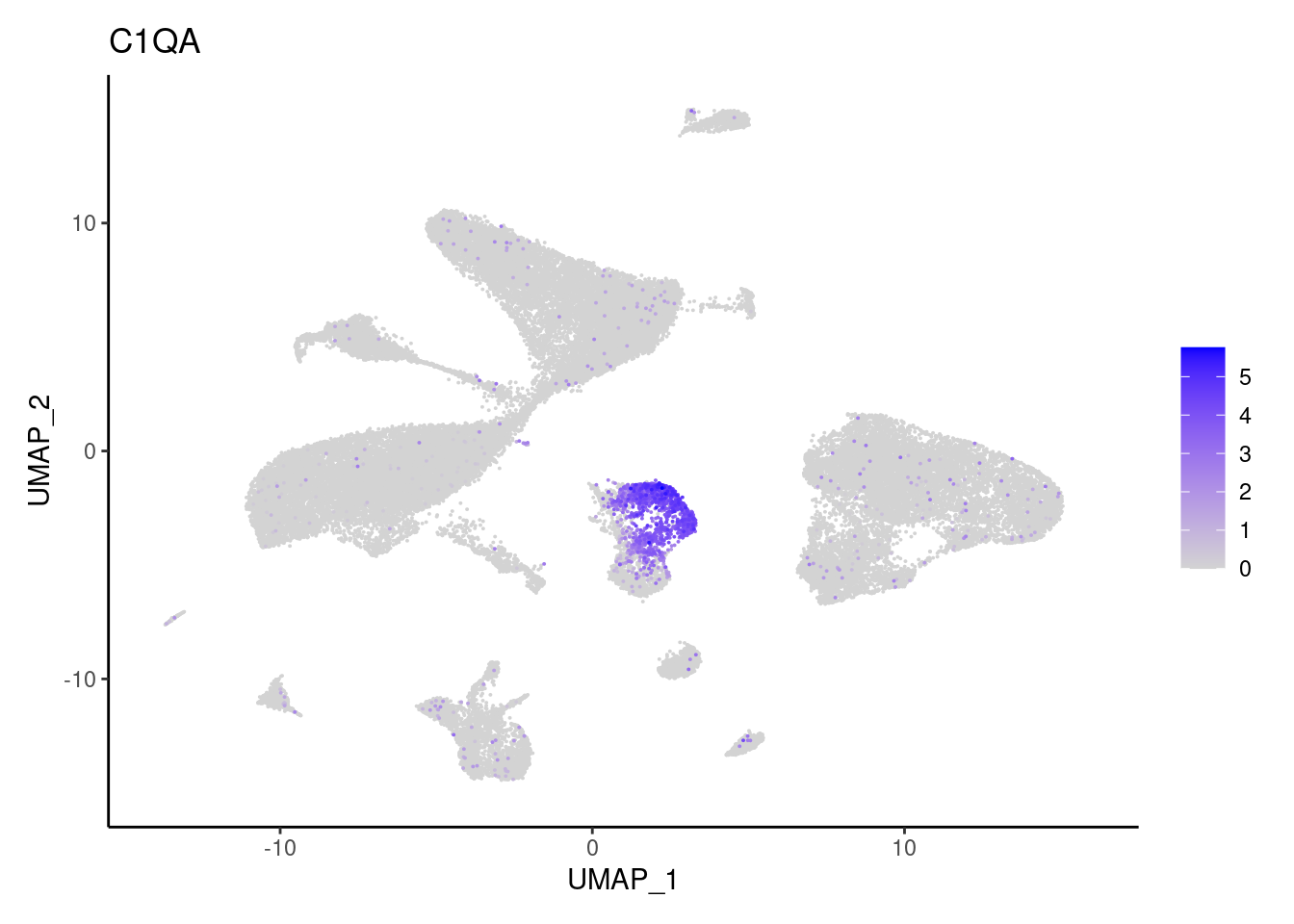
C1QB
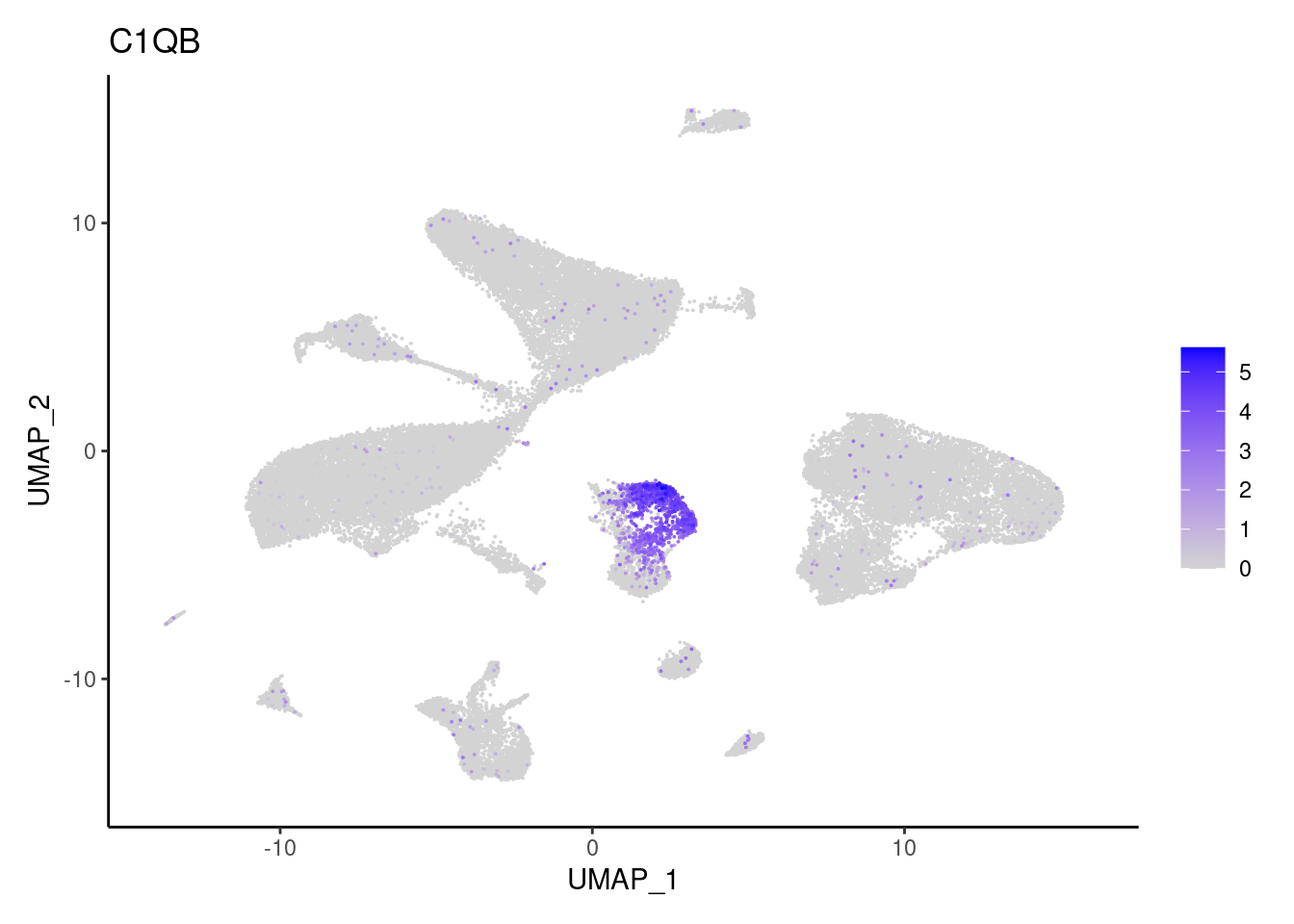
CD14
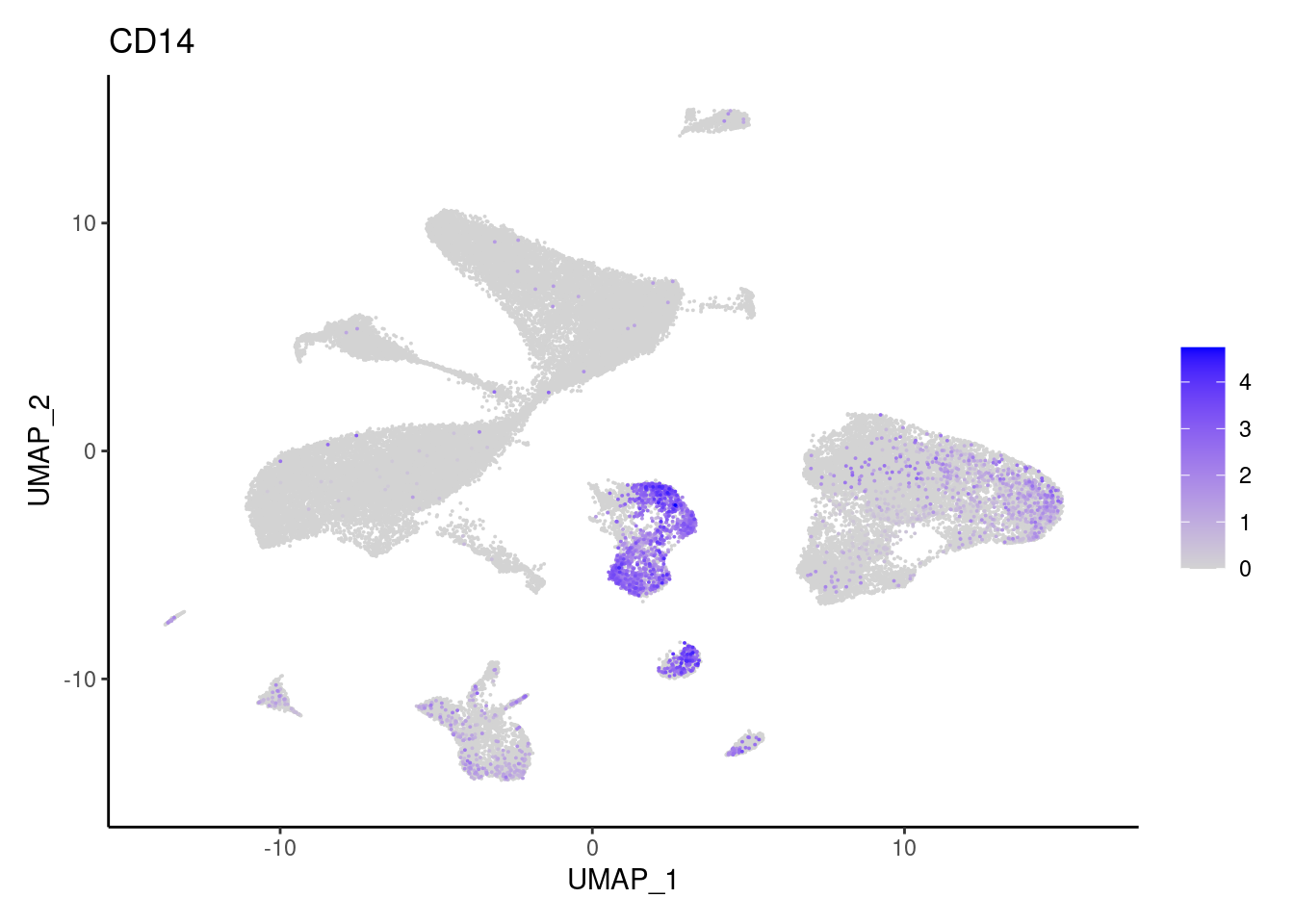
CMTM2
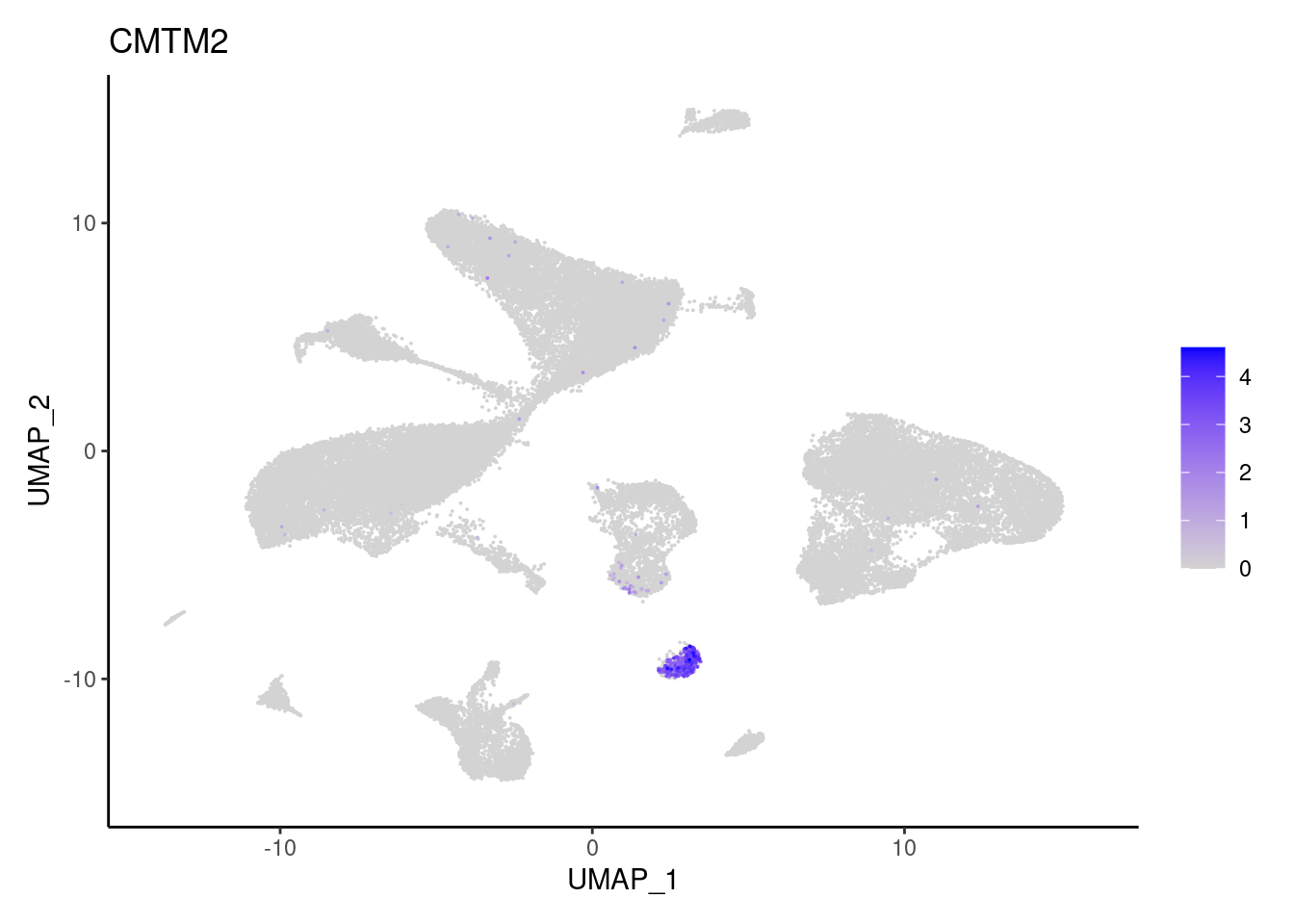
FCGR3B
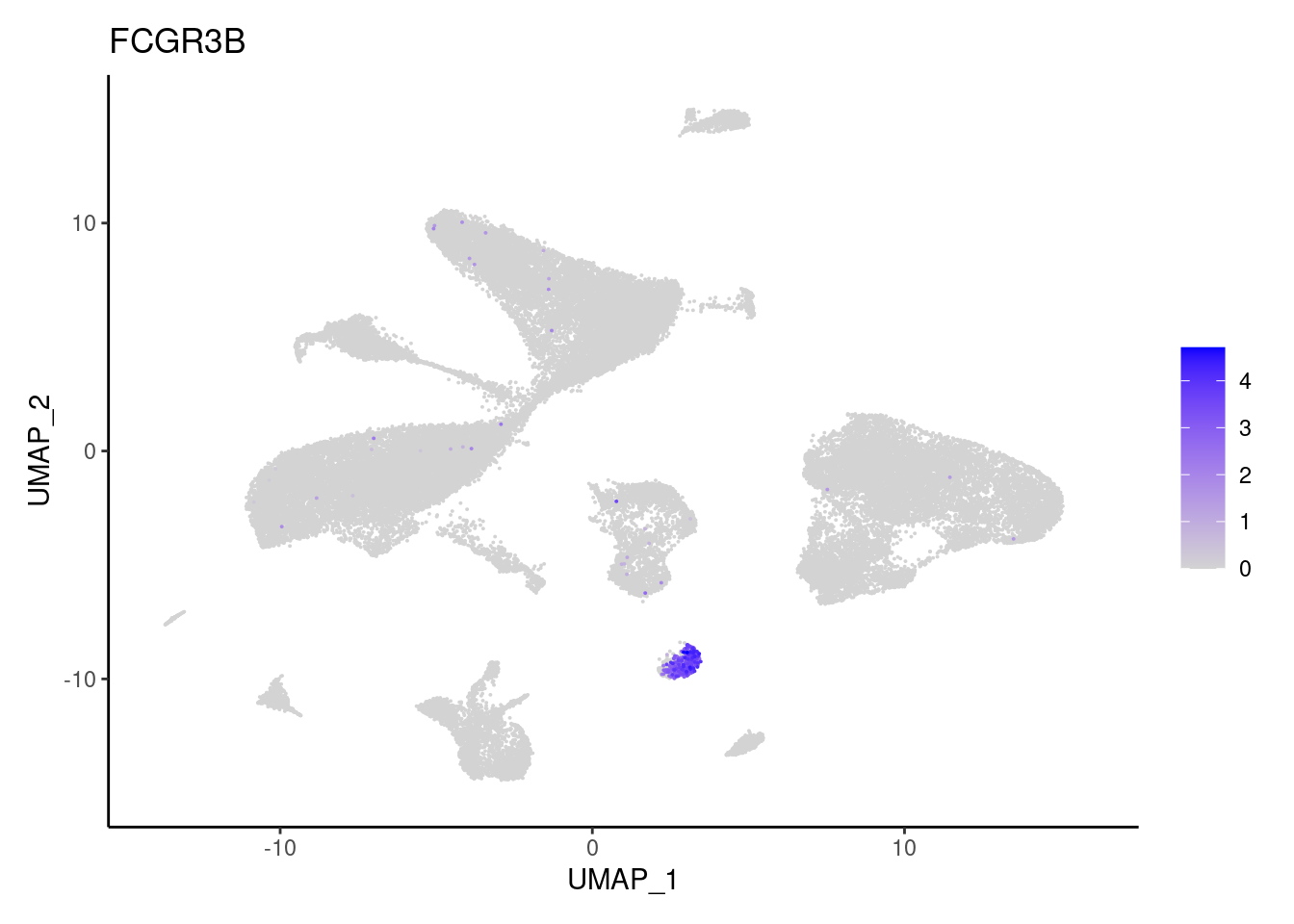
LYZ
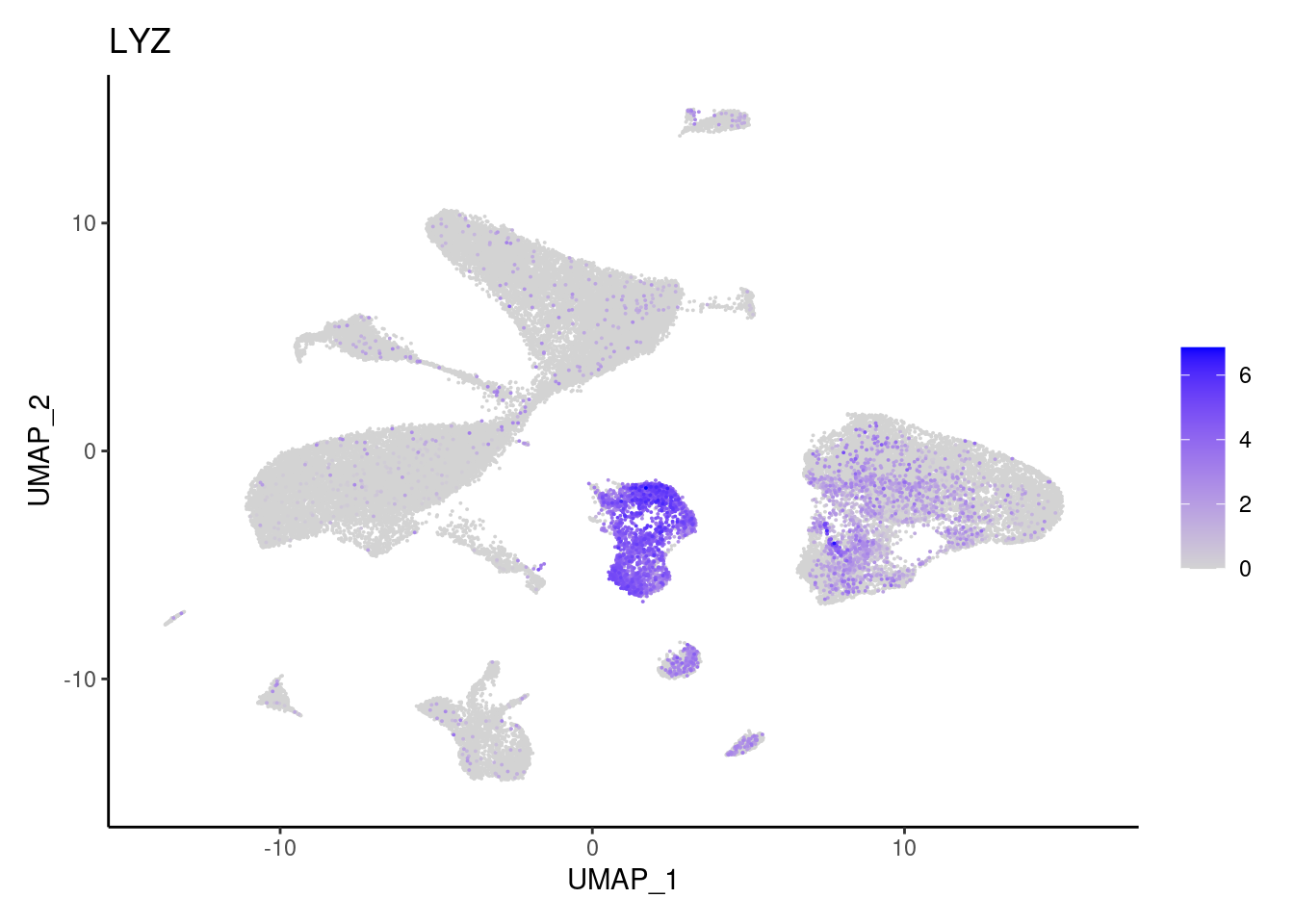
MS4A2
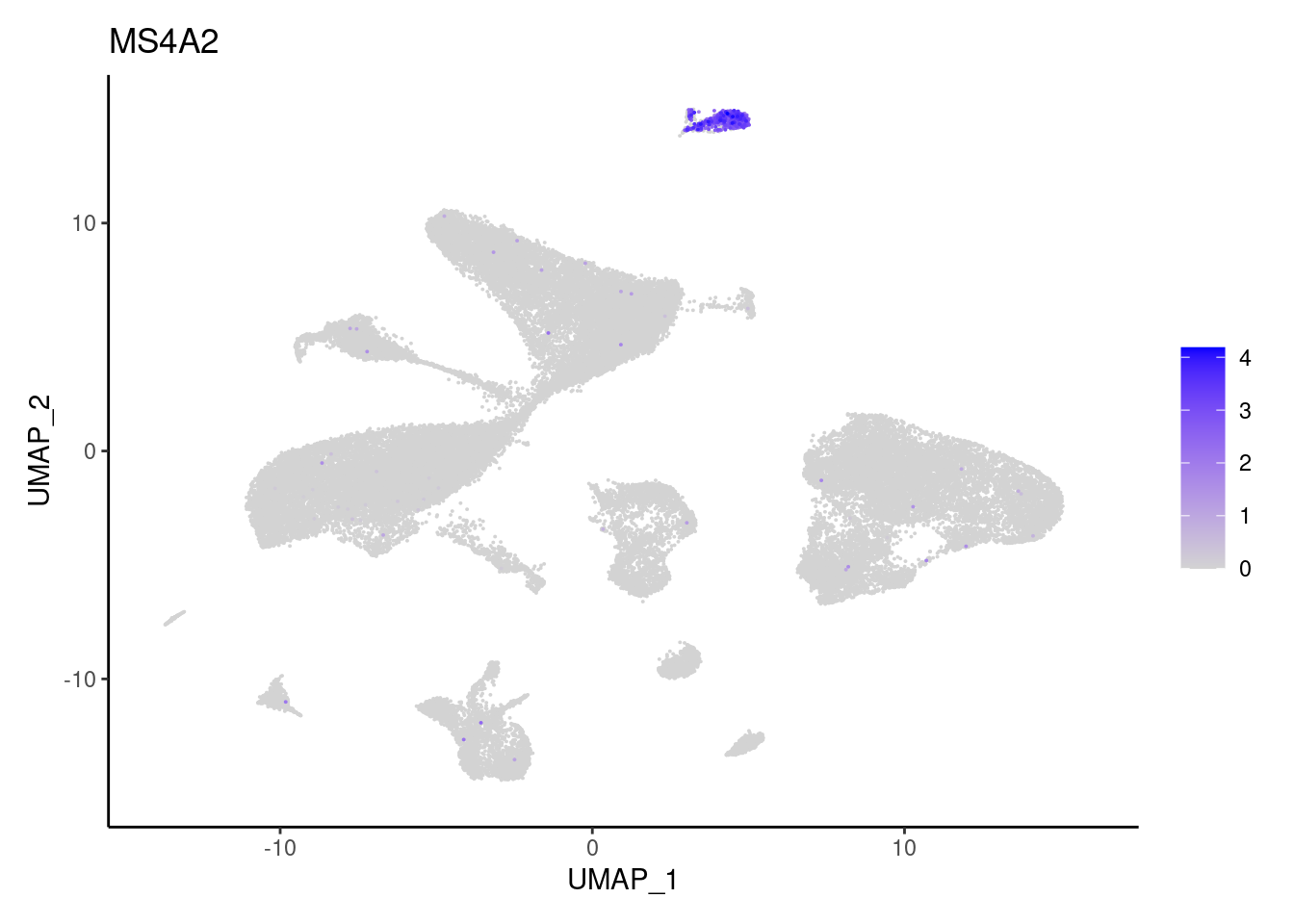
TPSAB1
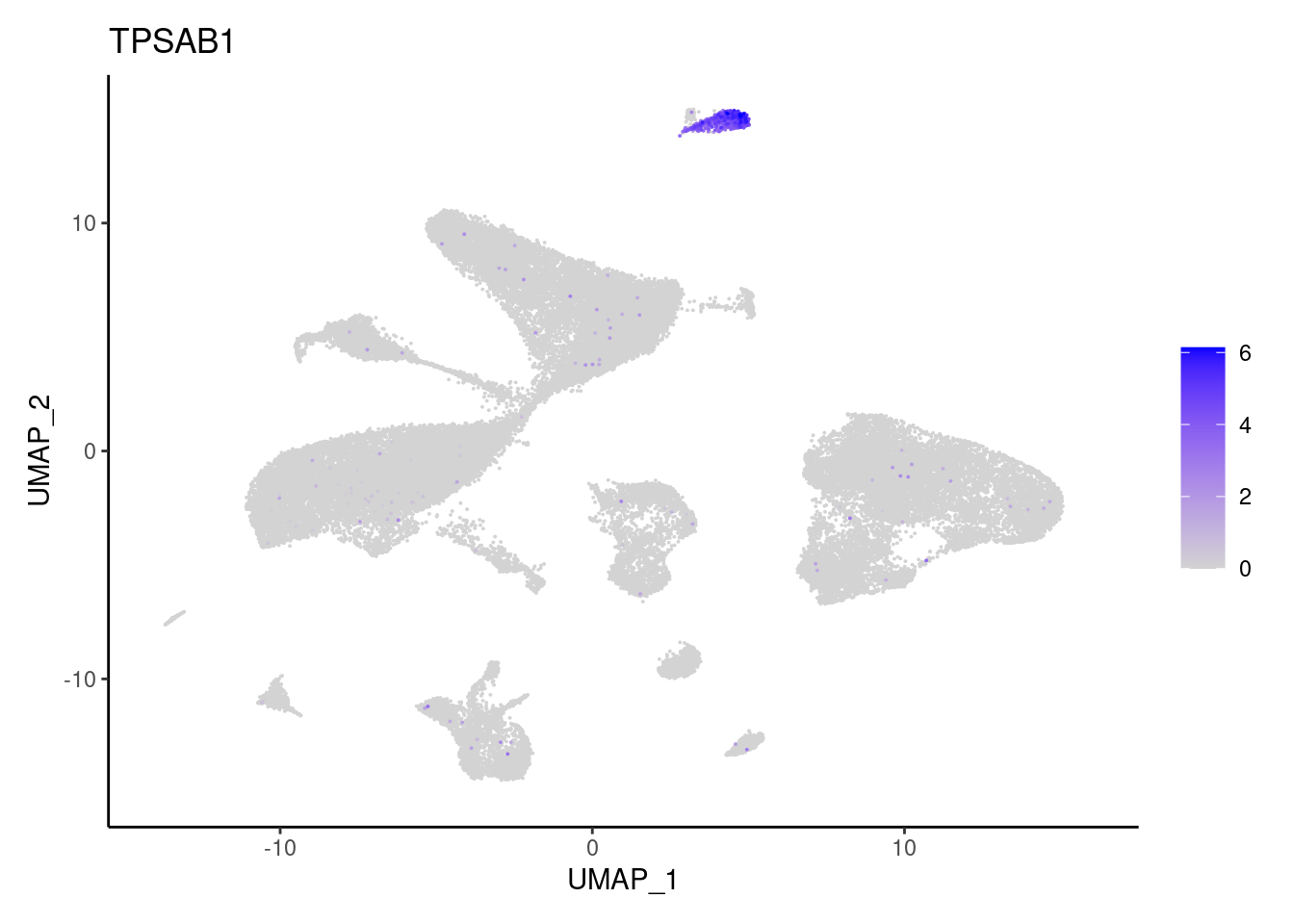
TPSB2
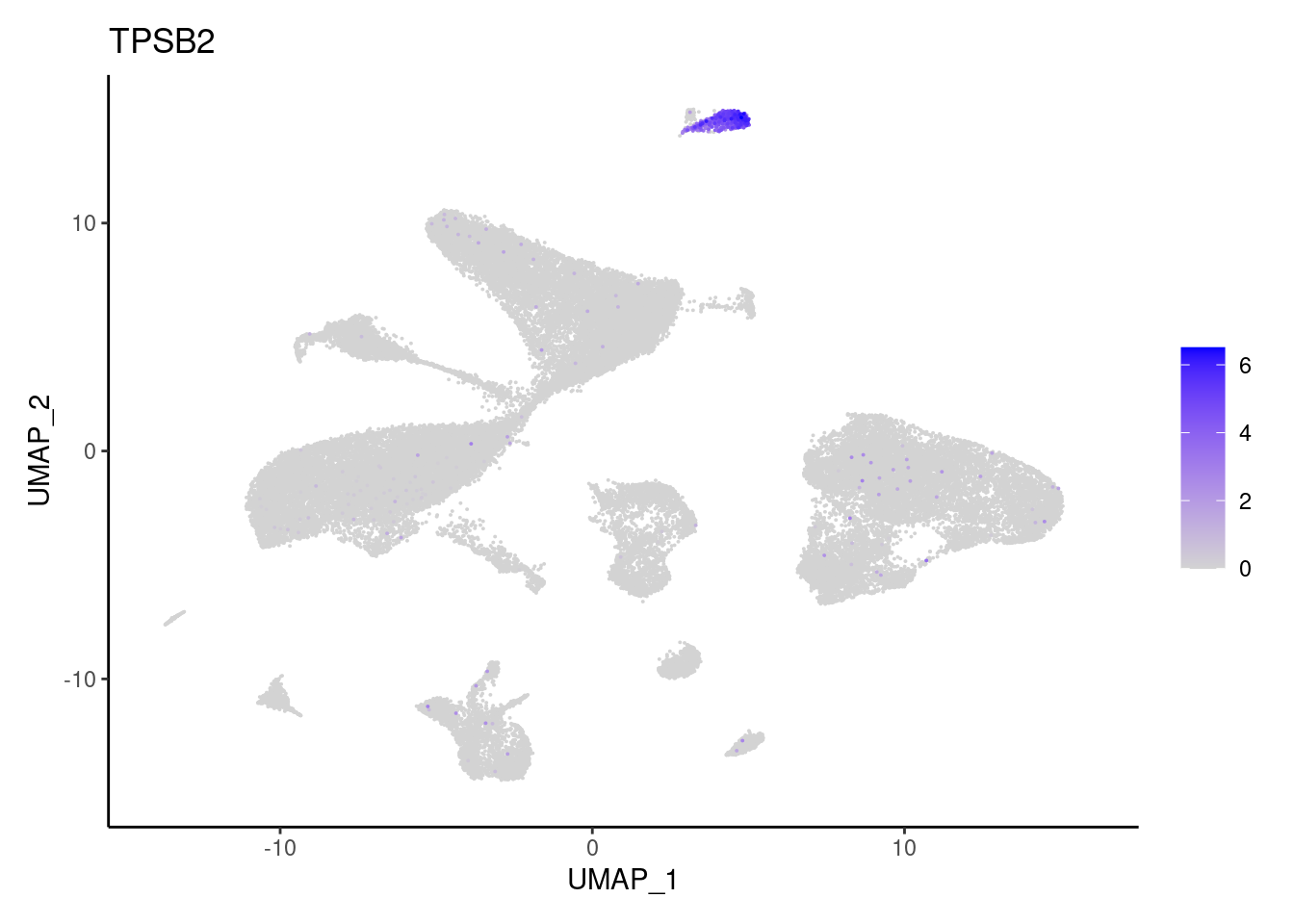
CPA3
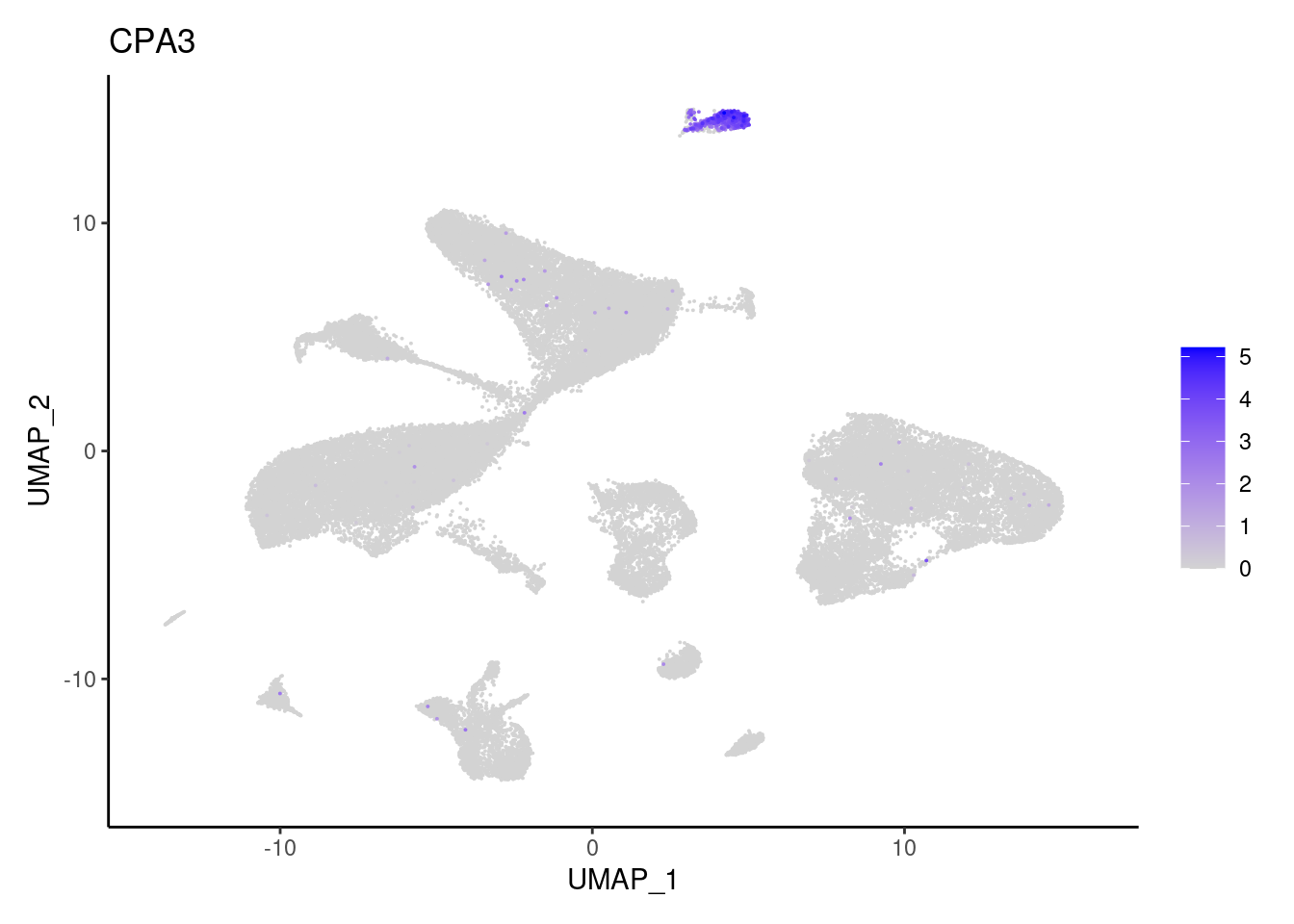
CD14
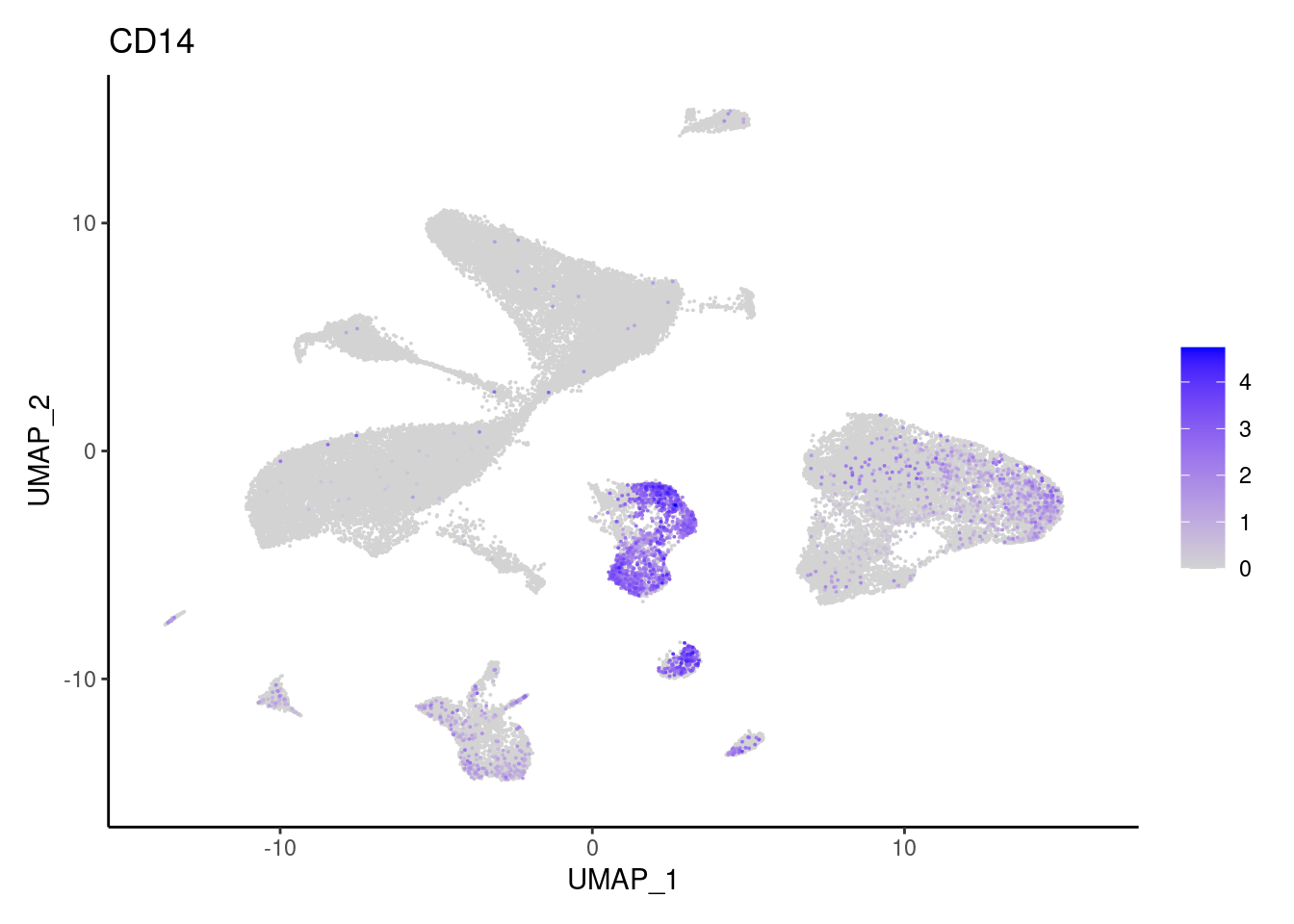
CD163
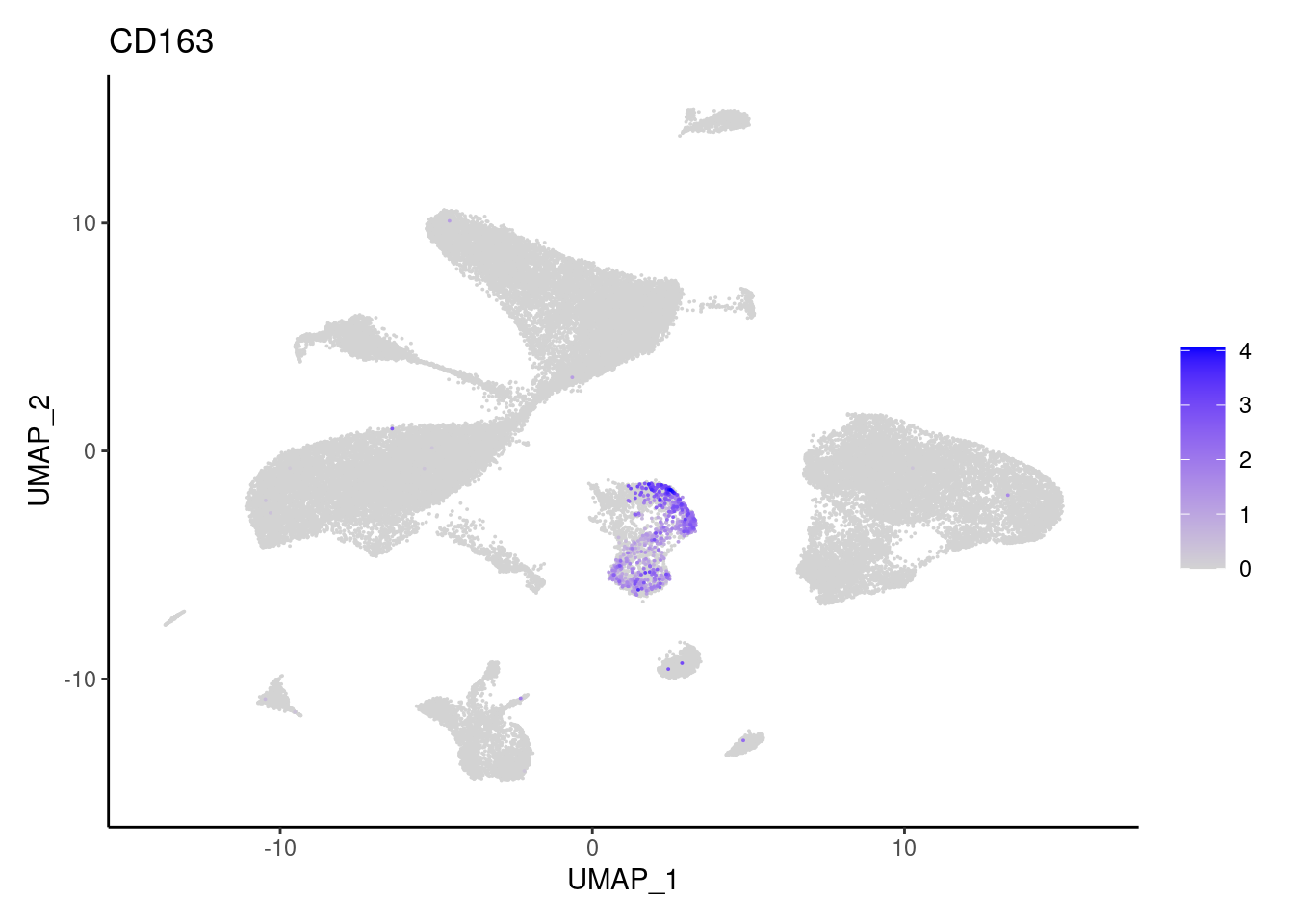
CSF1R
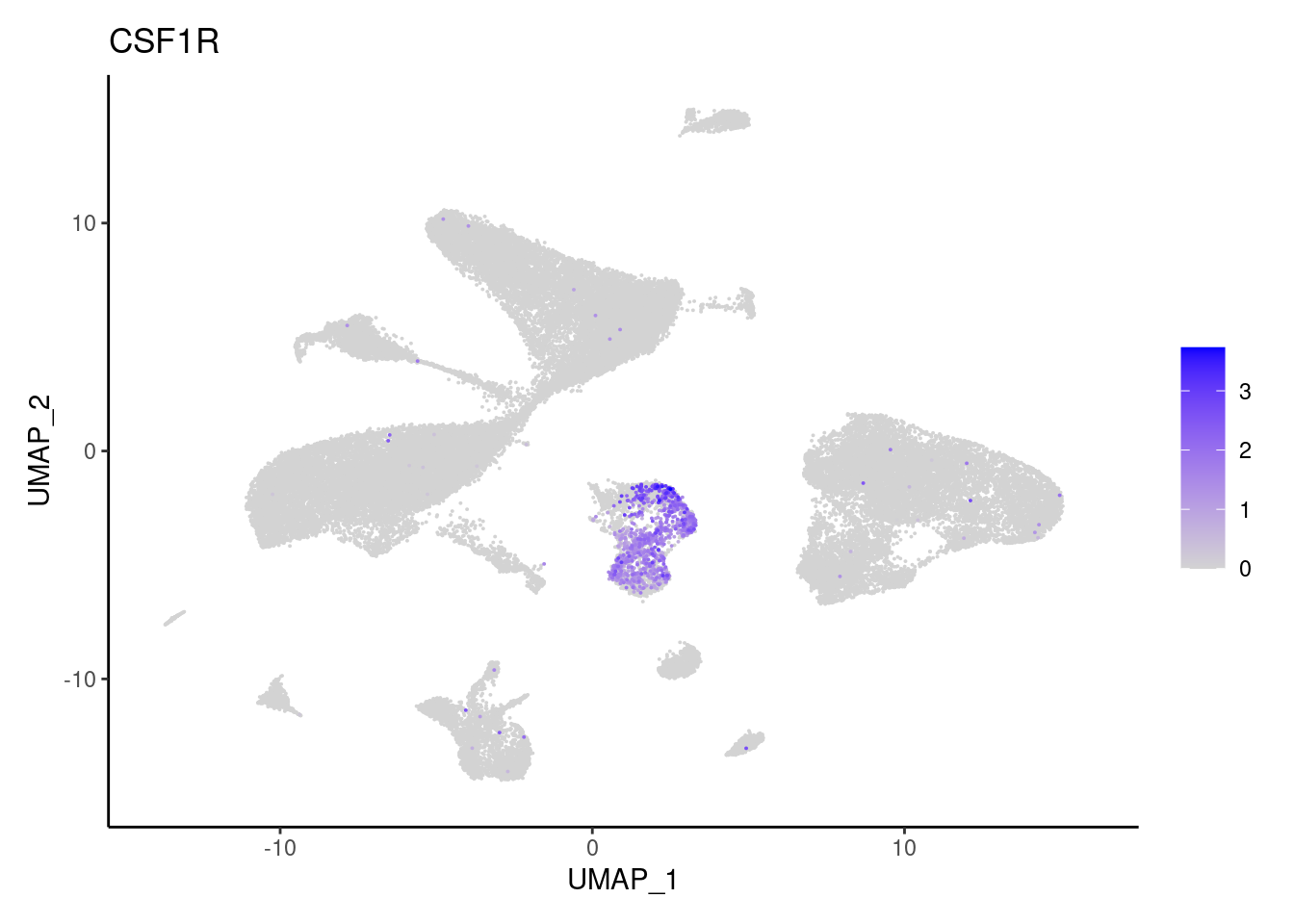
CD68
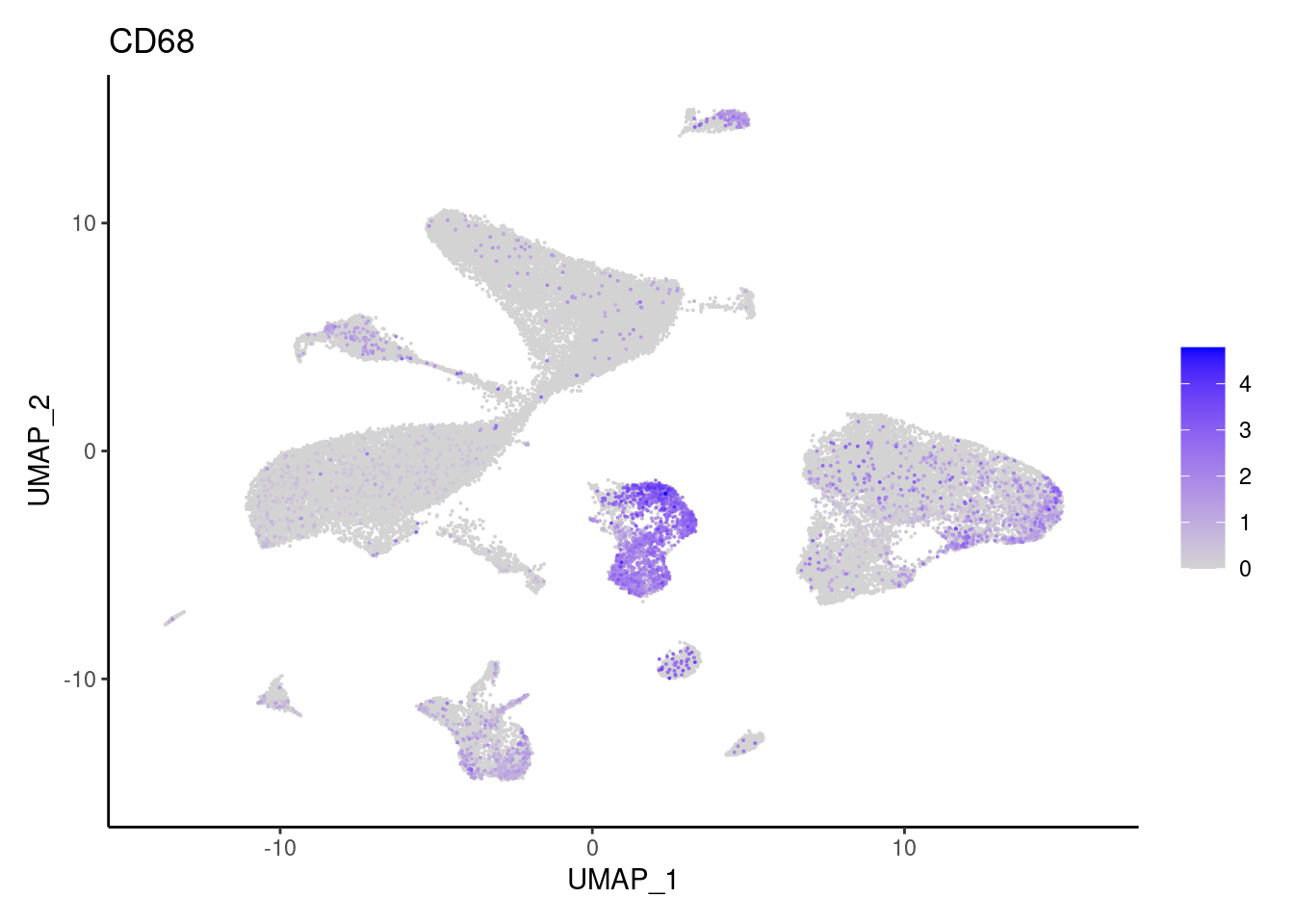
Stromal cells
ACTA2
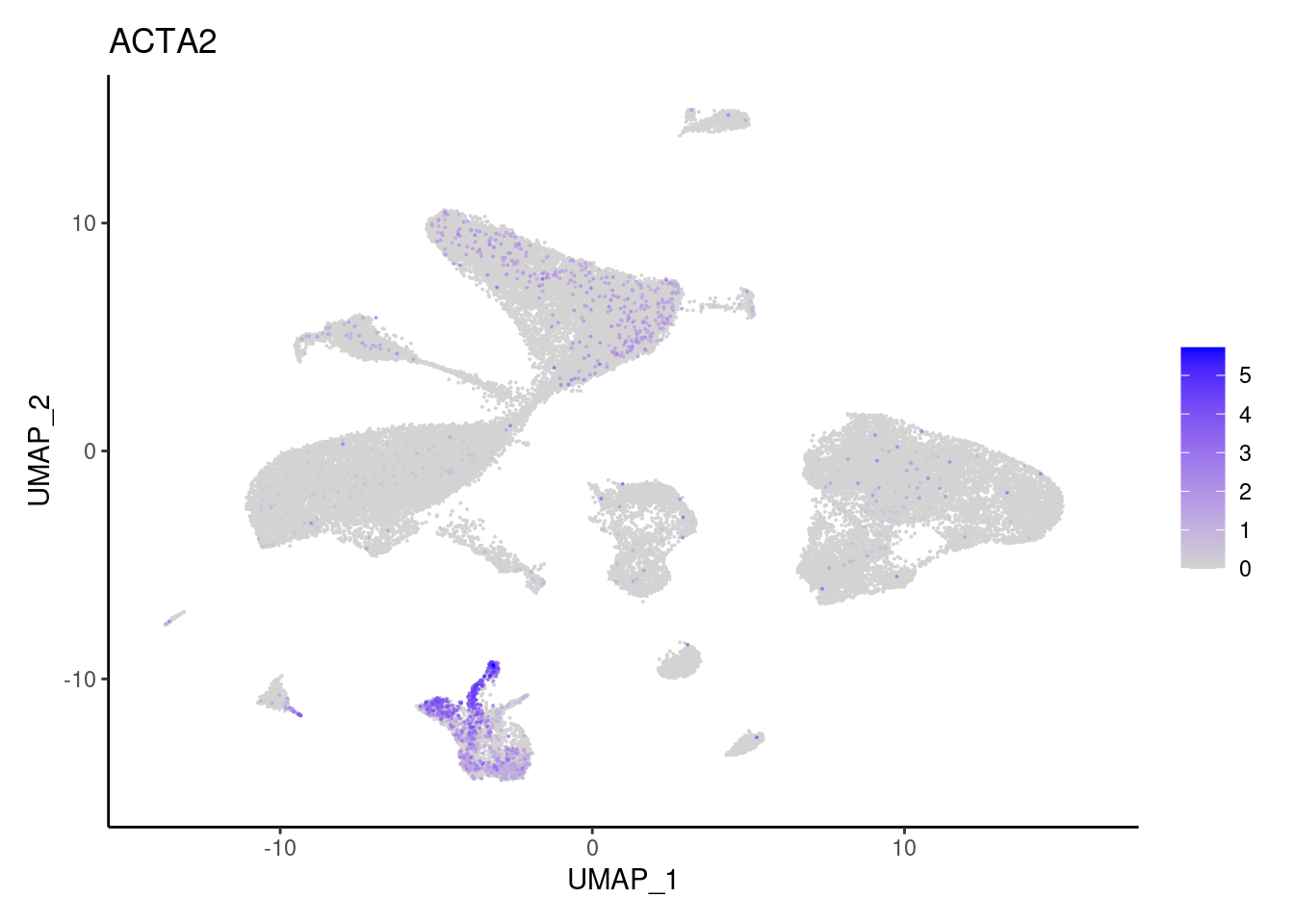
ADAMDEC1
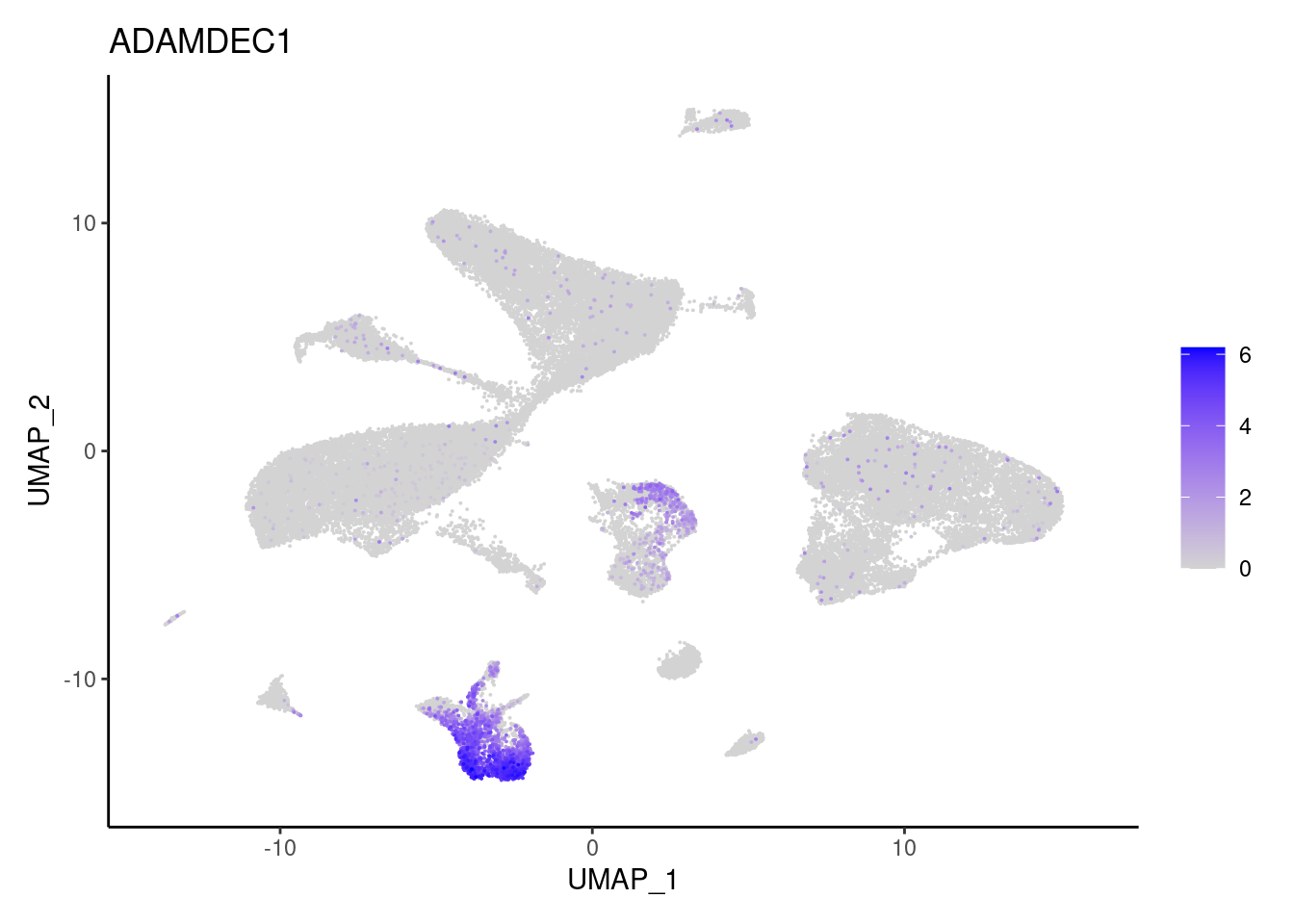
CHI3L1
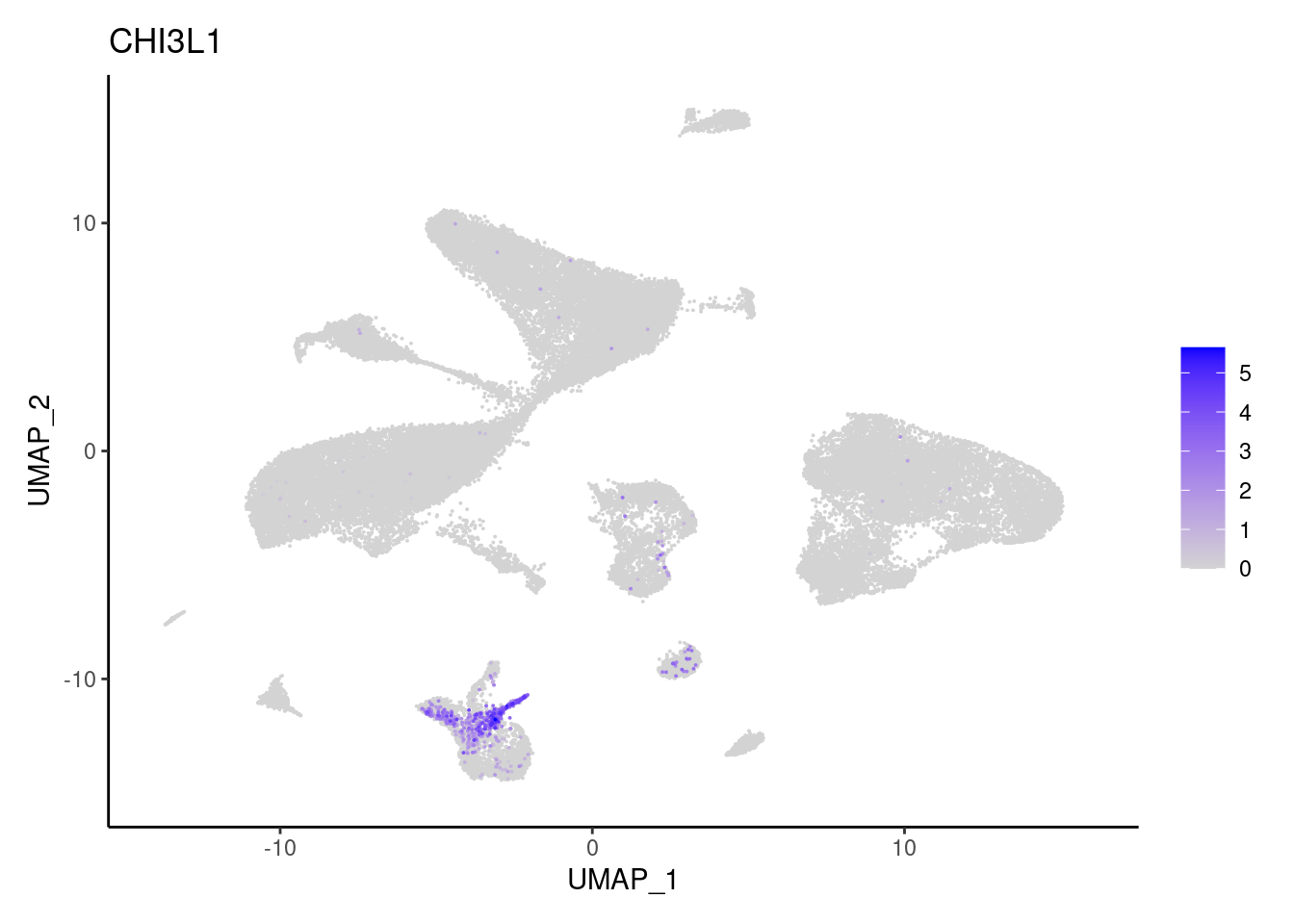
COL3A1
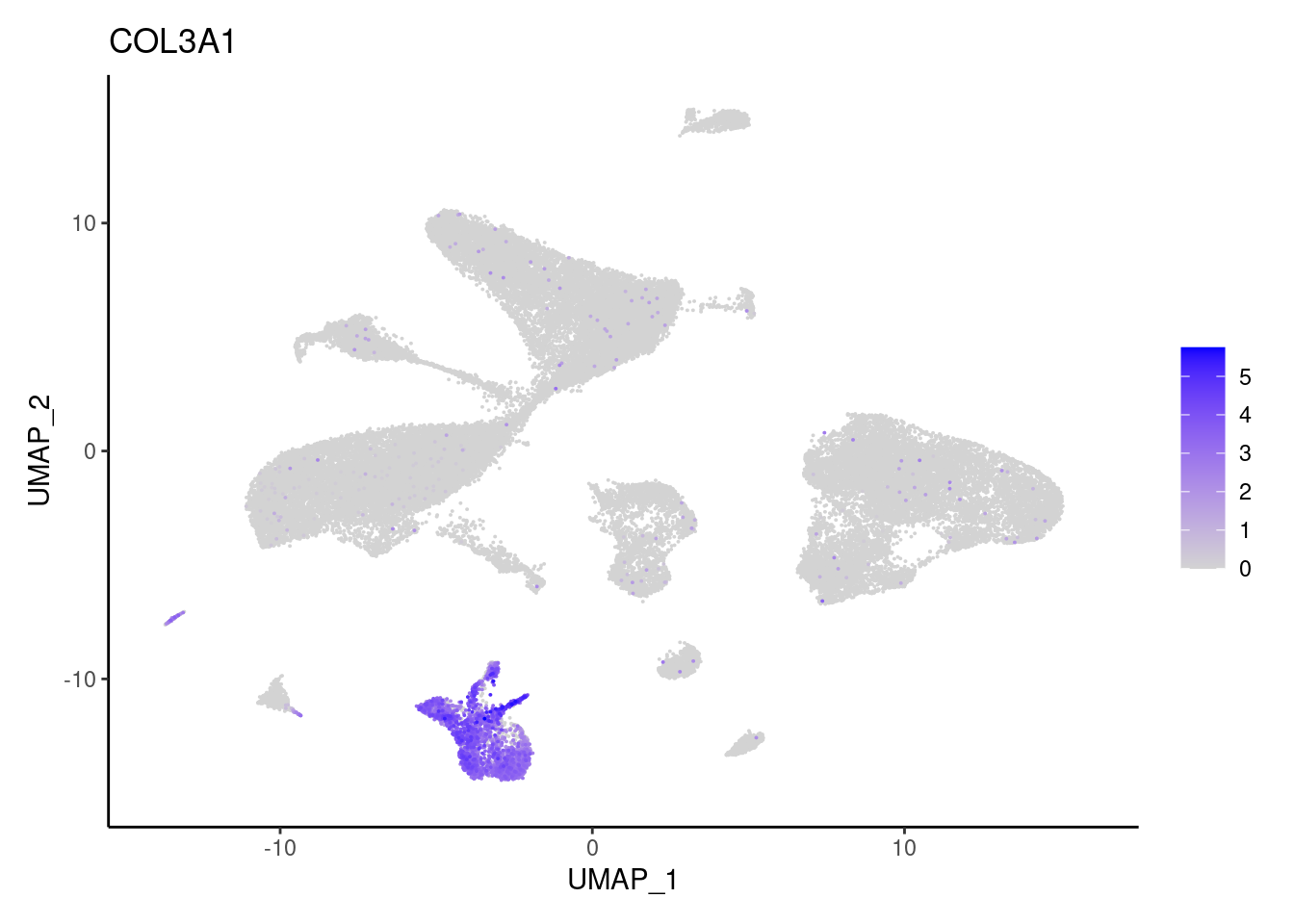
NRXN1
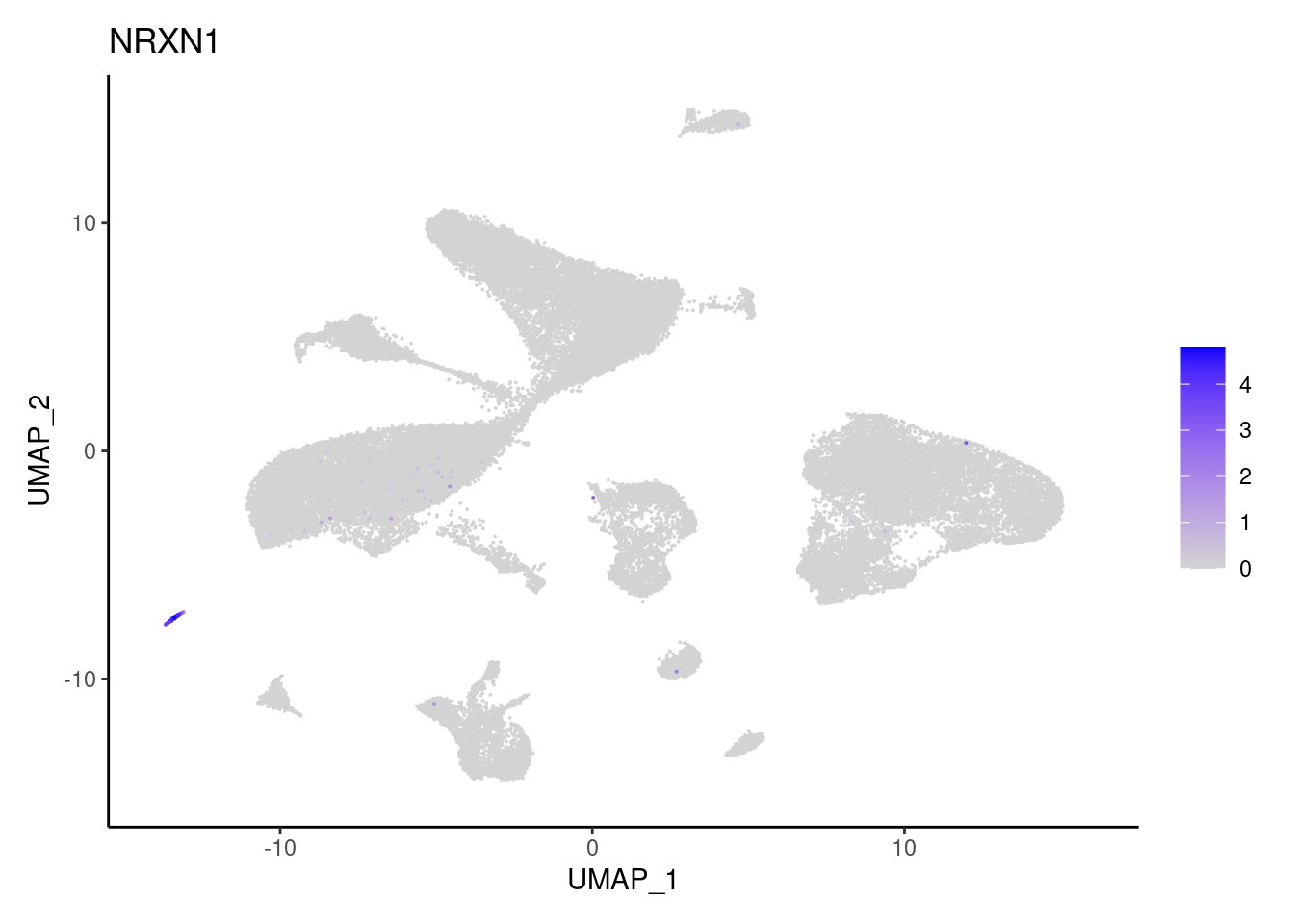
PLVAP
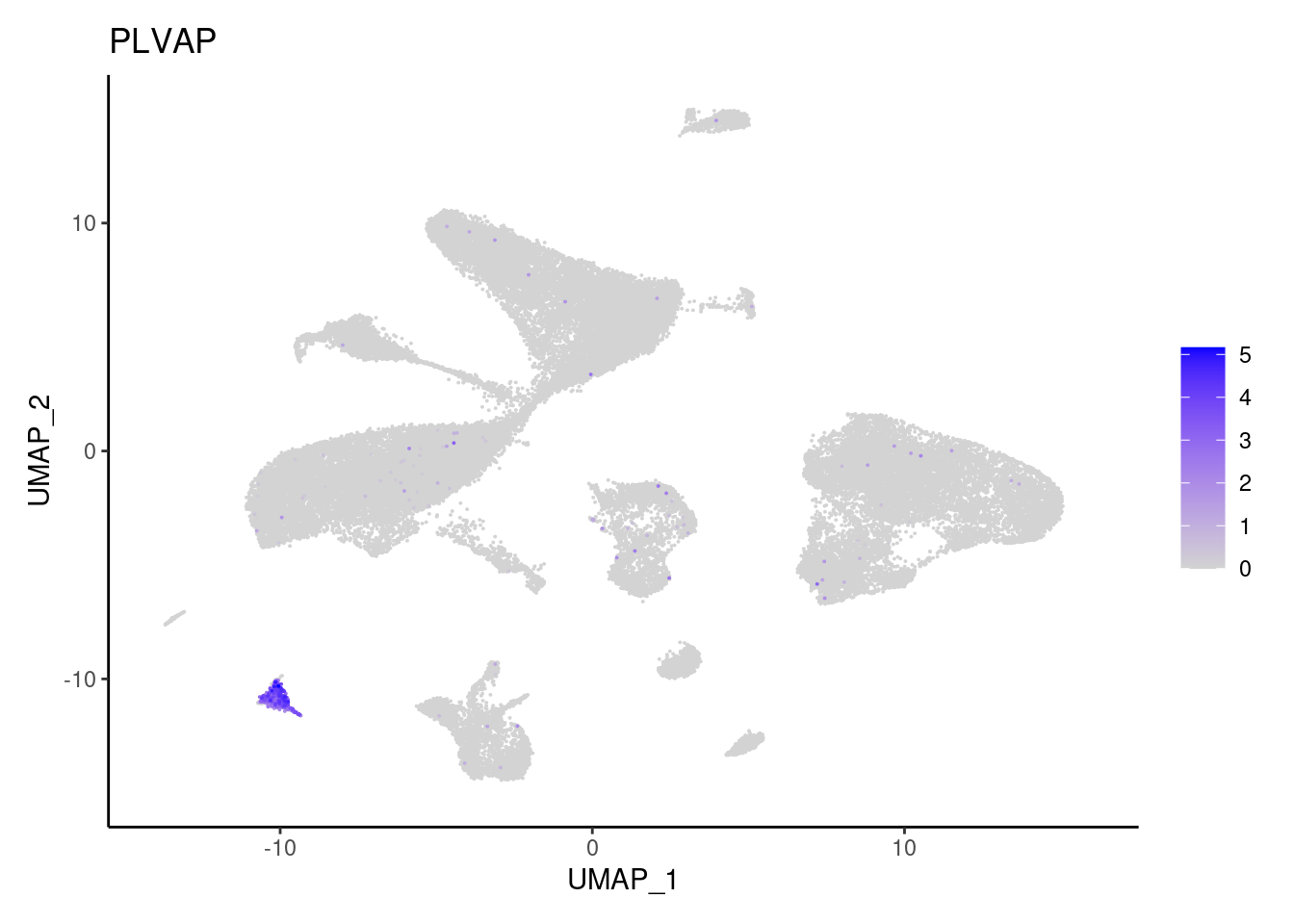
SOX6
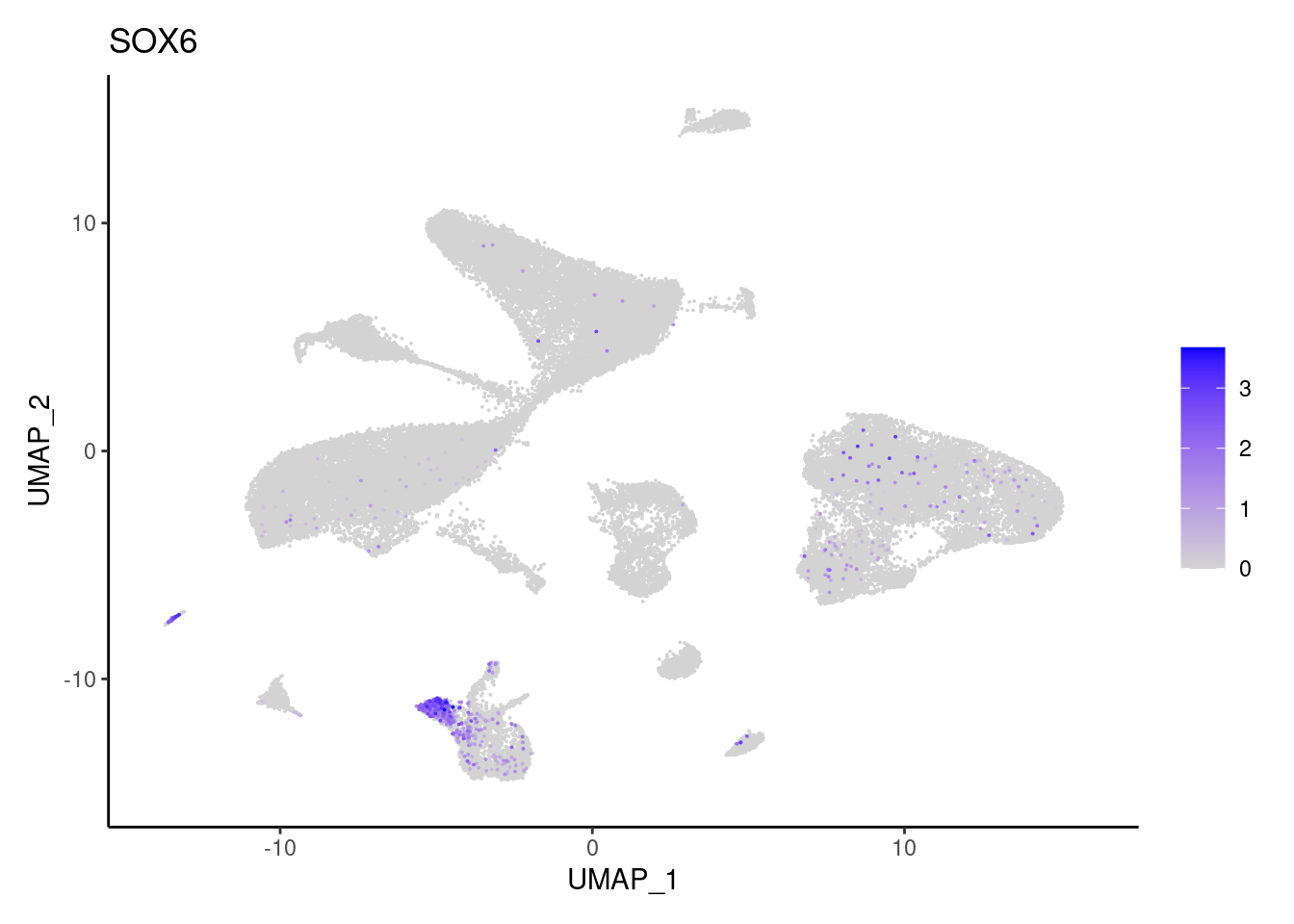
VWF
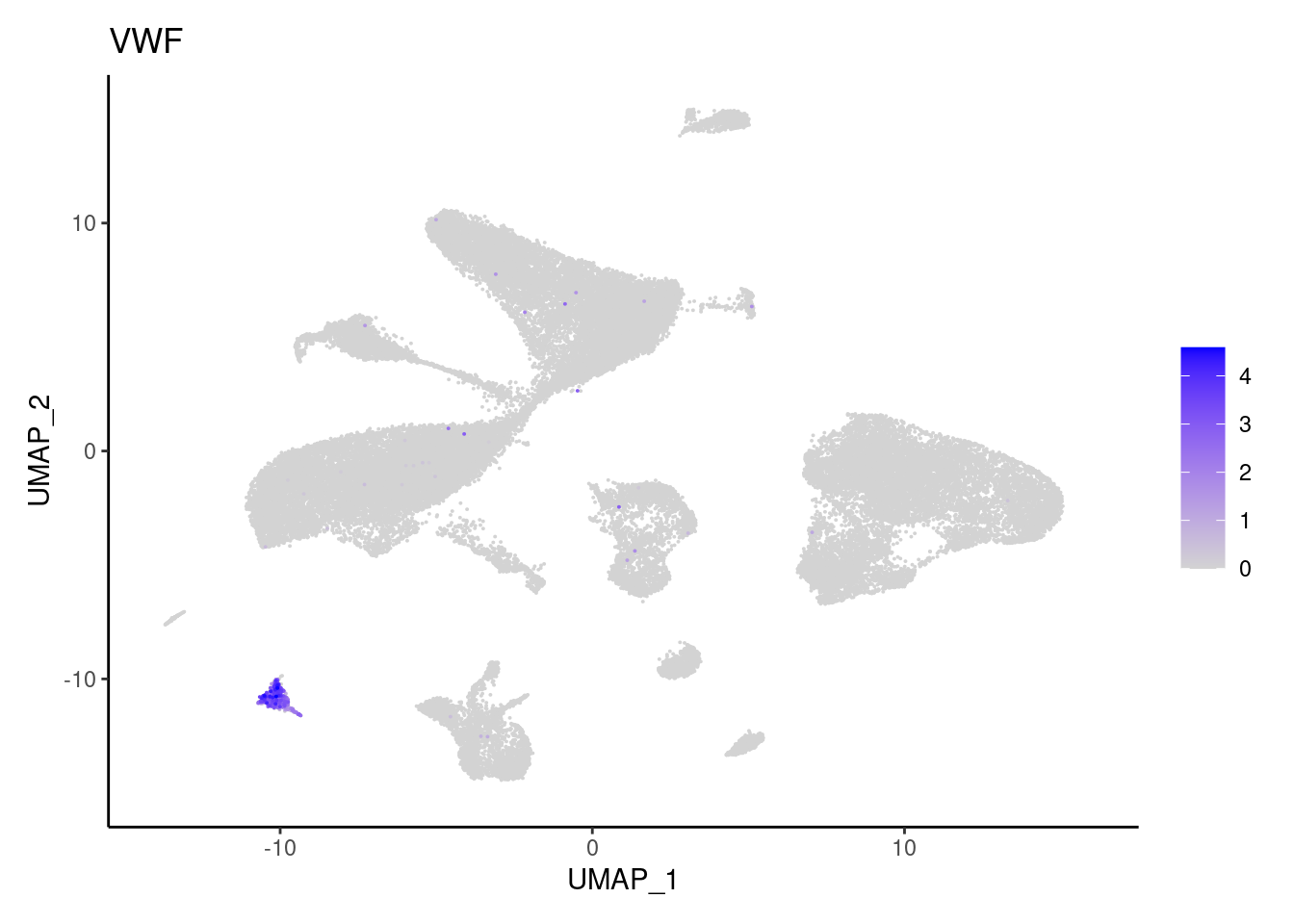
PDGFRA
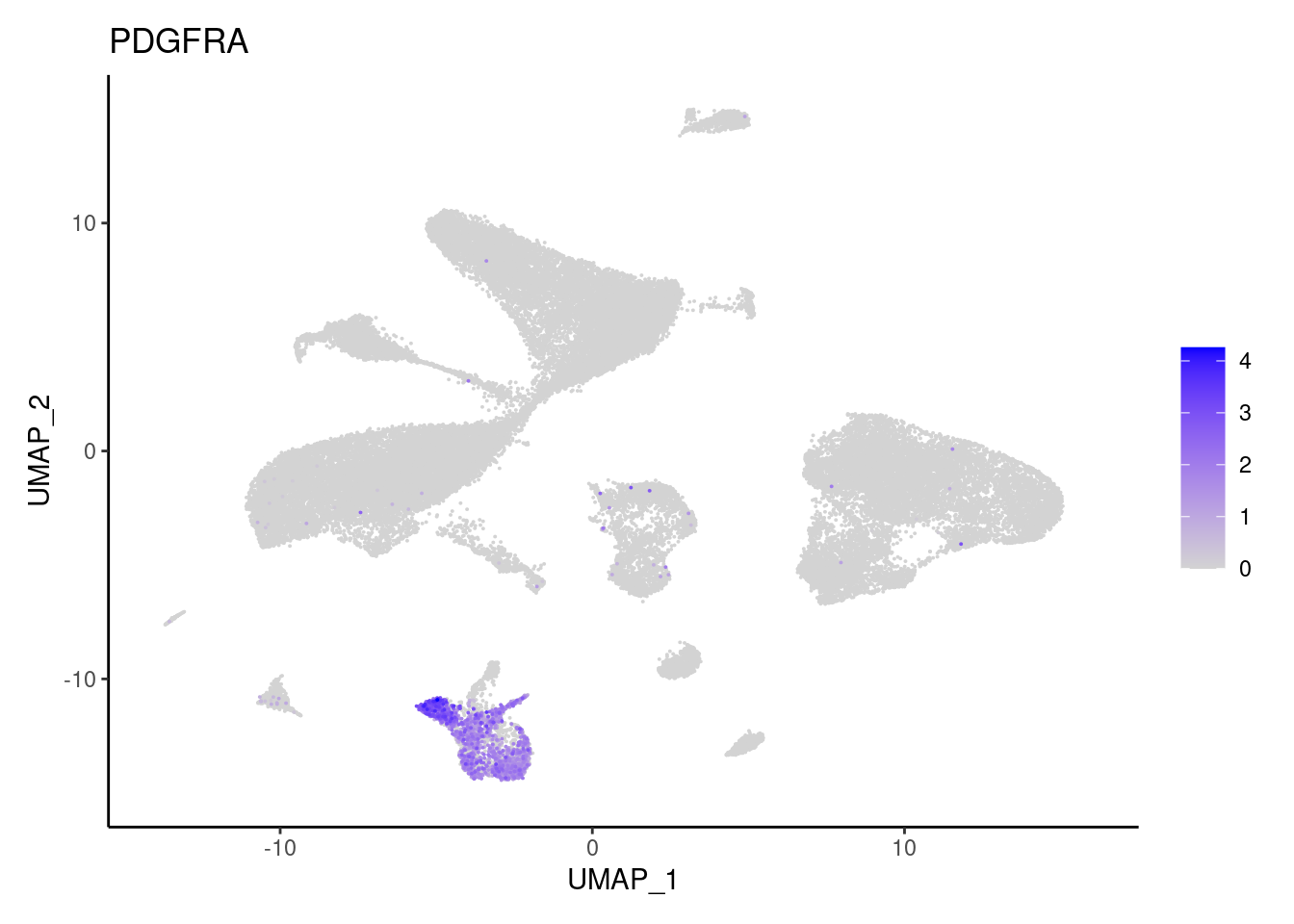
PDGFRB
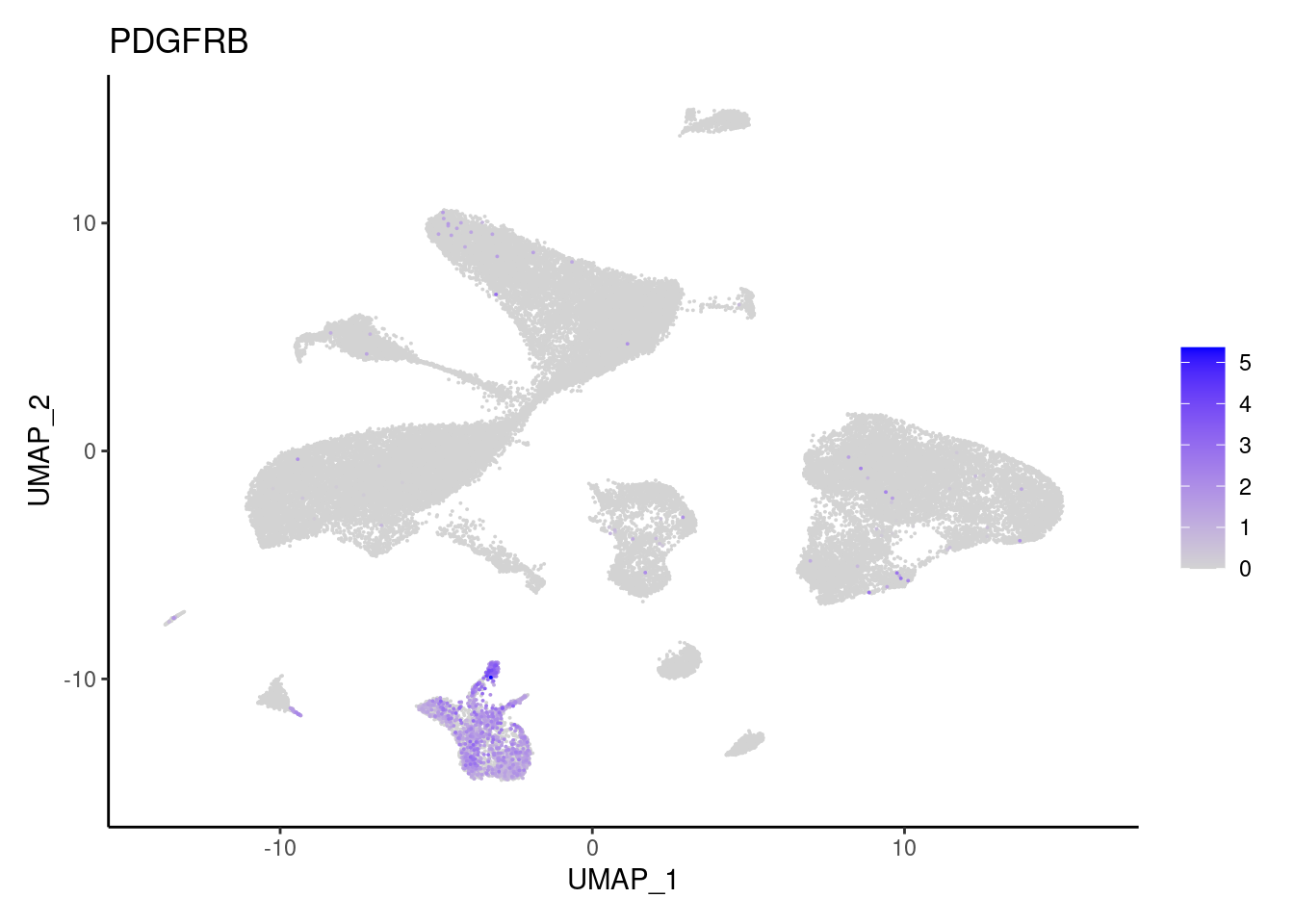
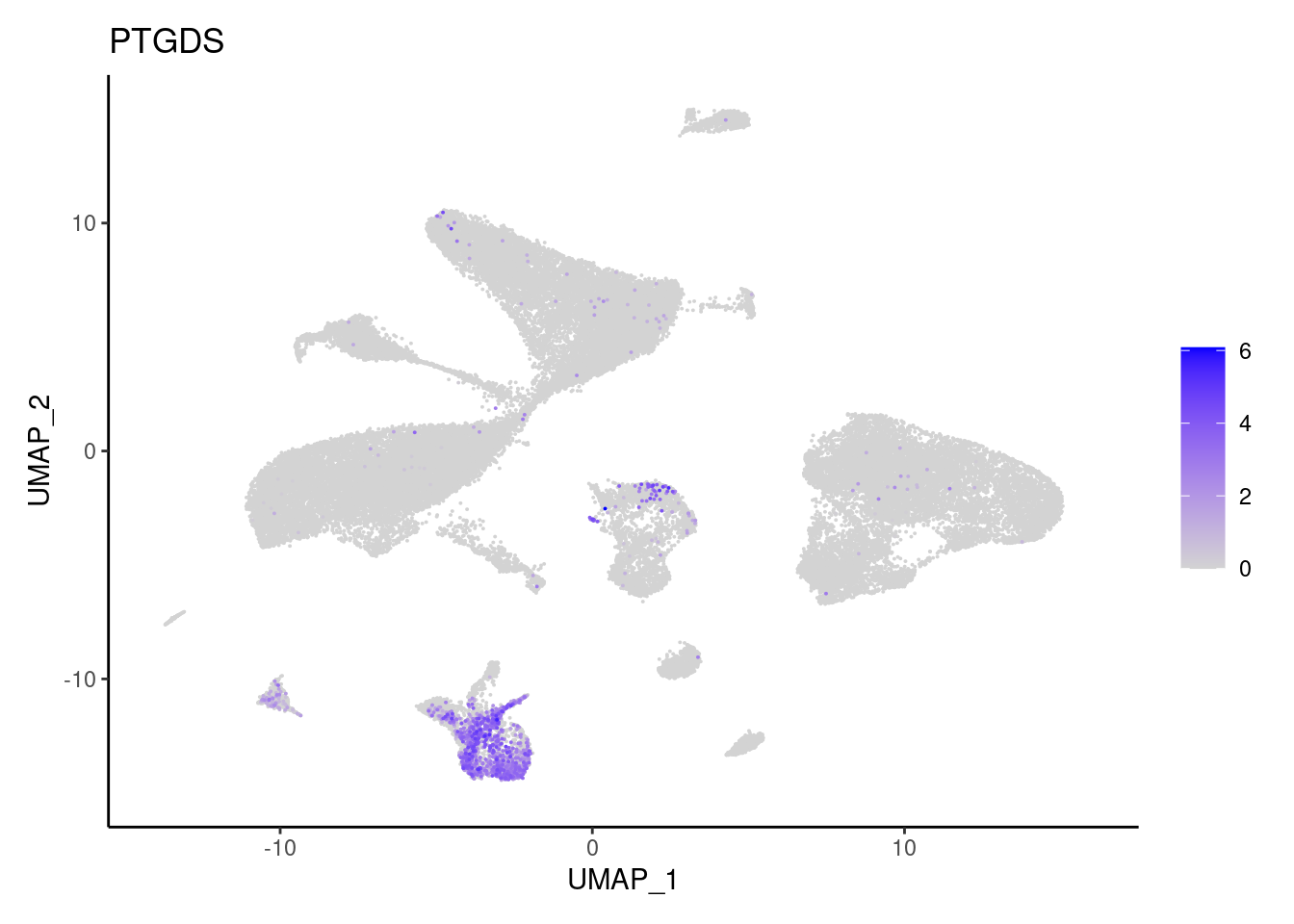
PTGDS
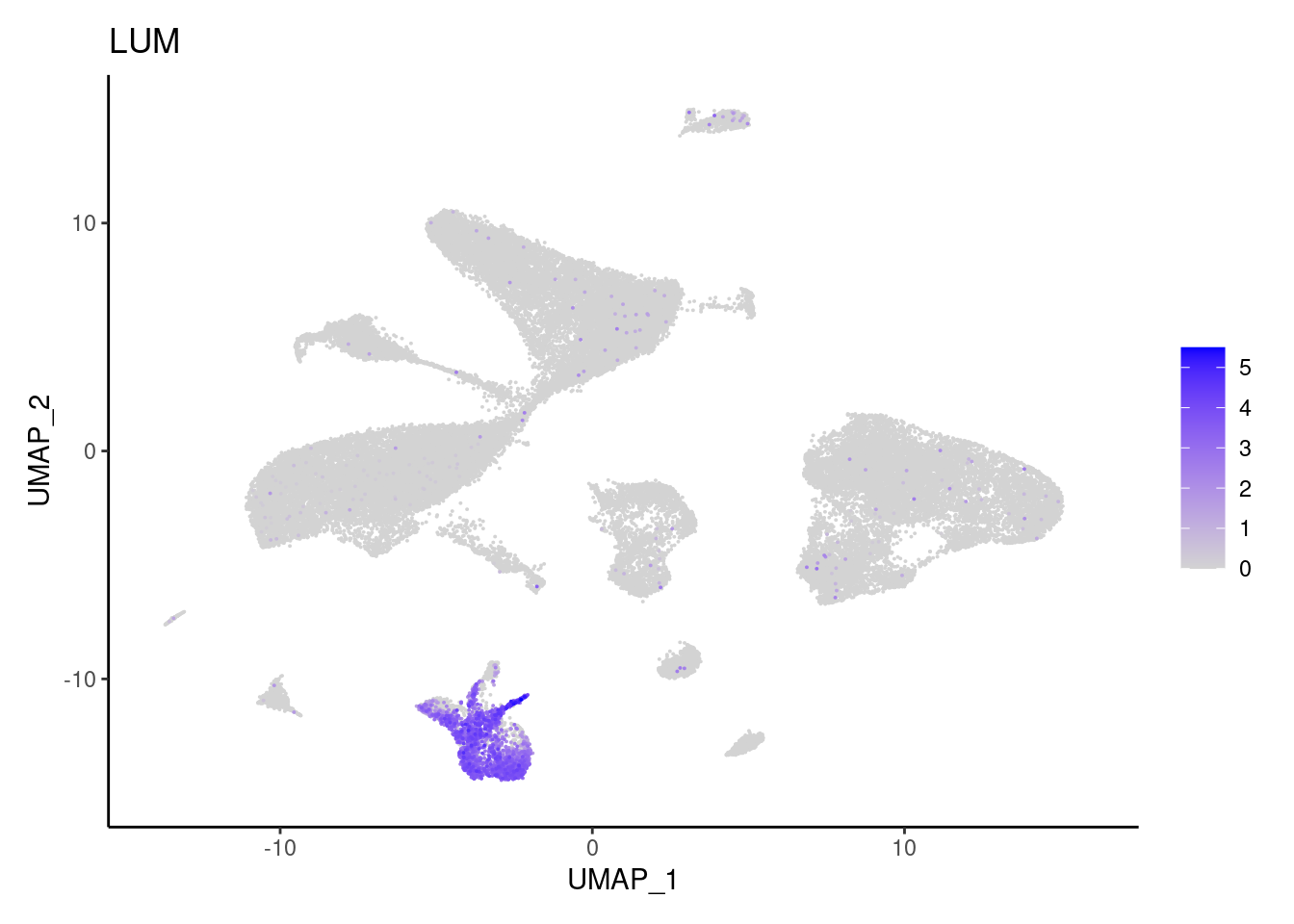
LUM
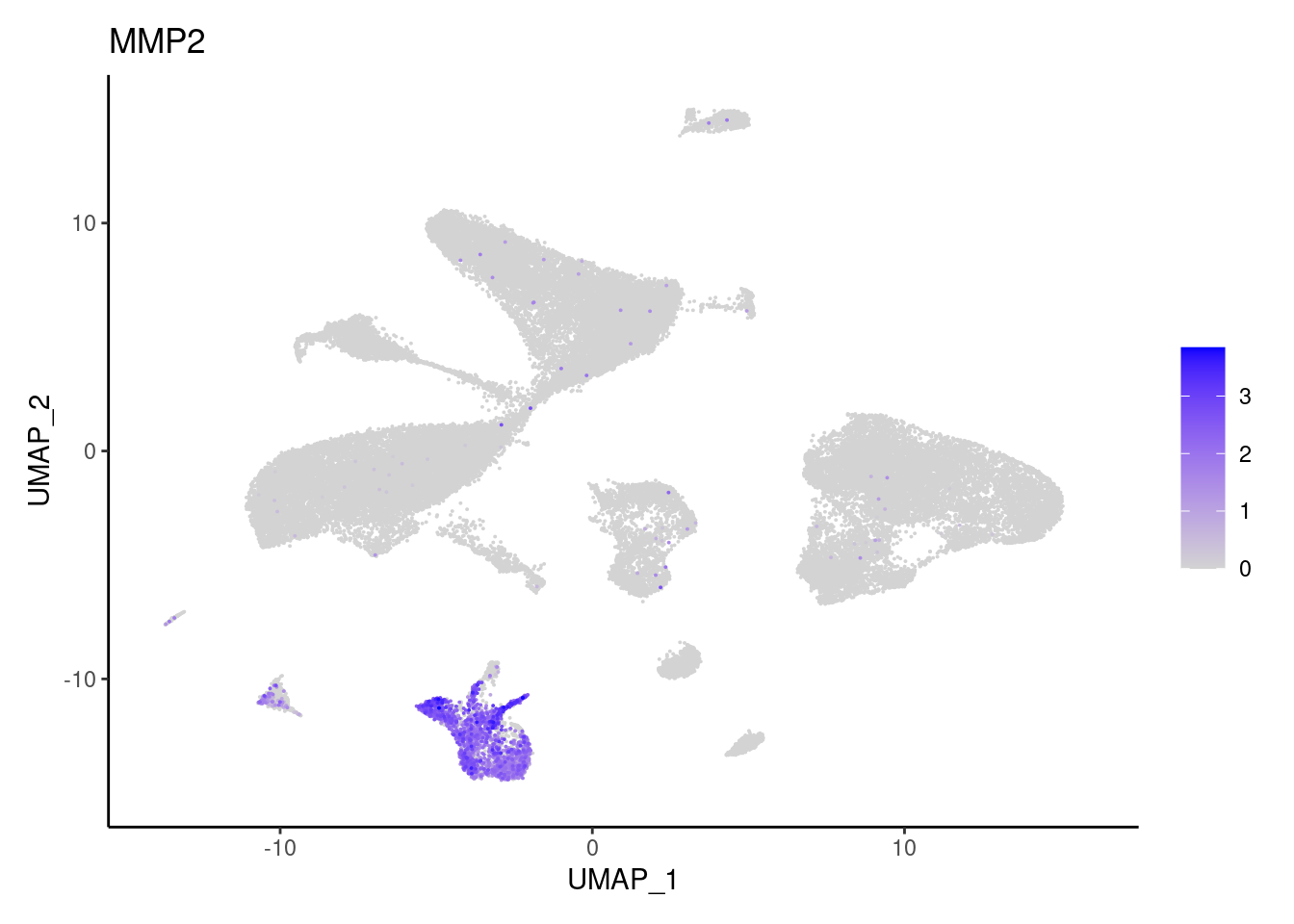
MMP2
T cells
CD3D
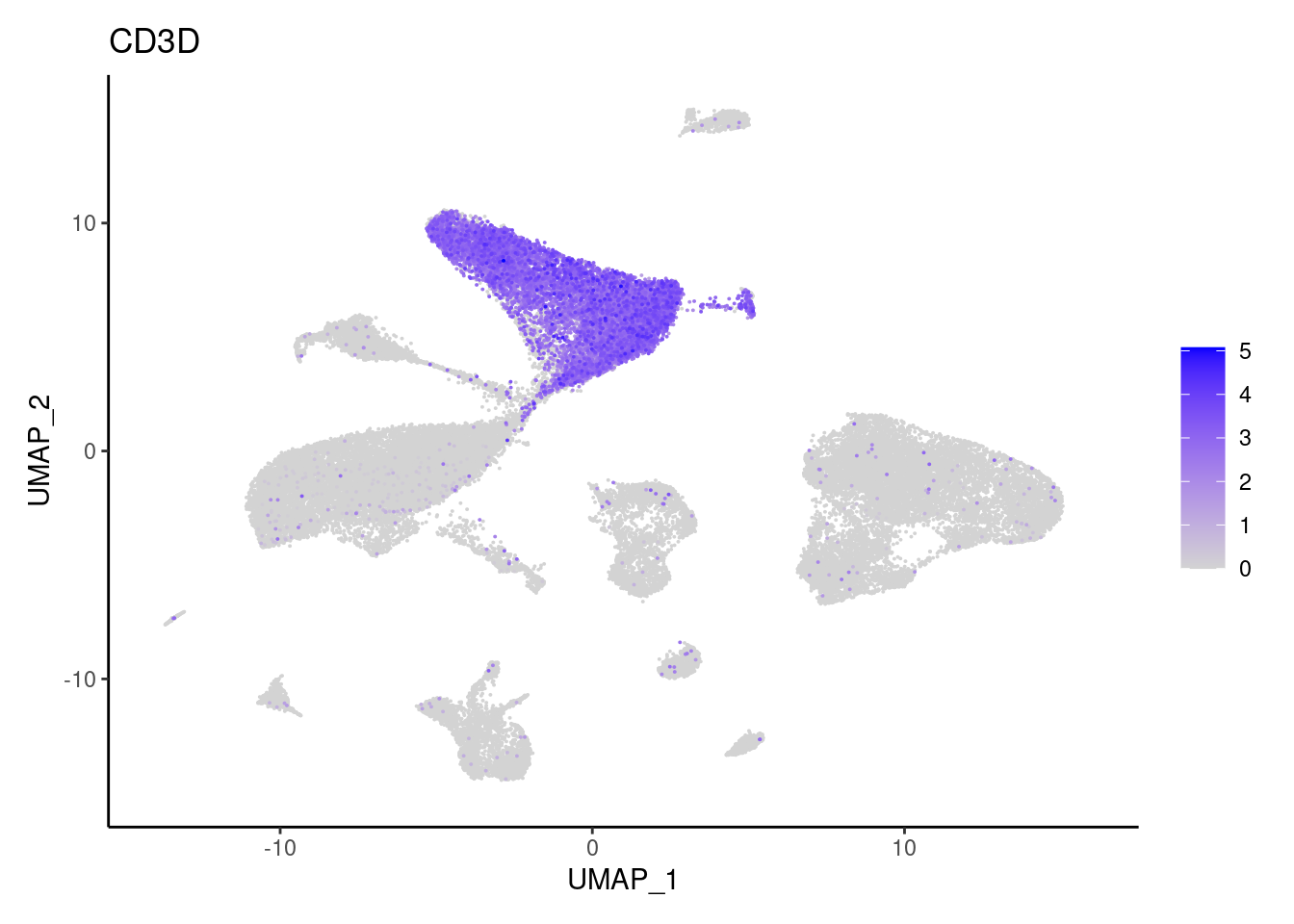
CD3E
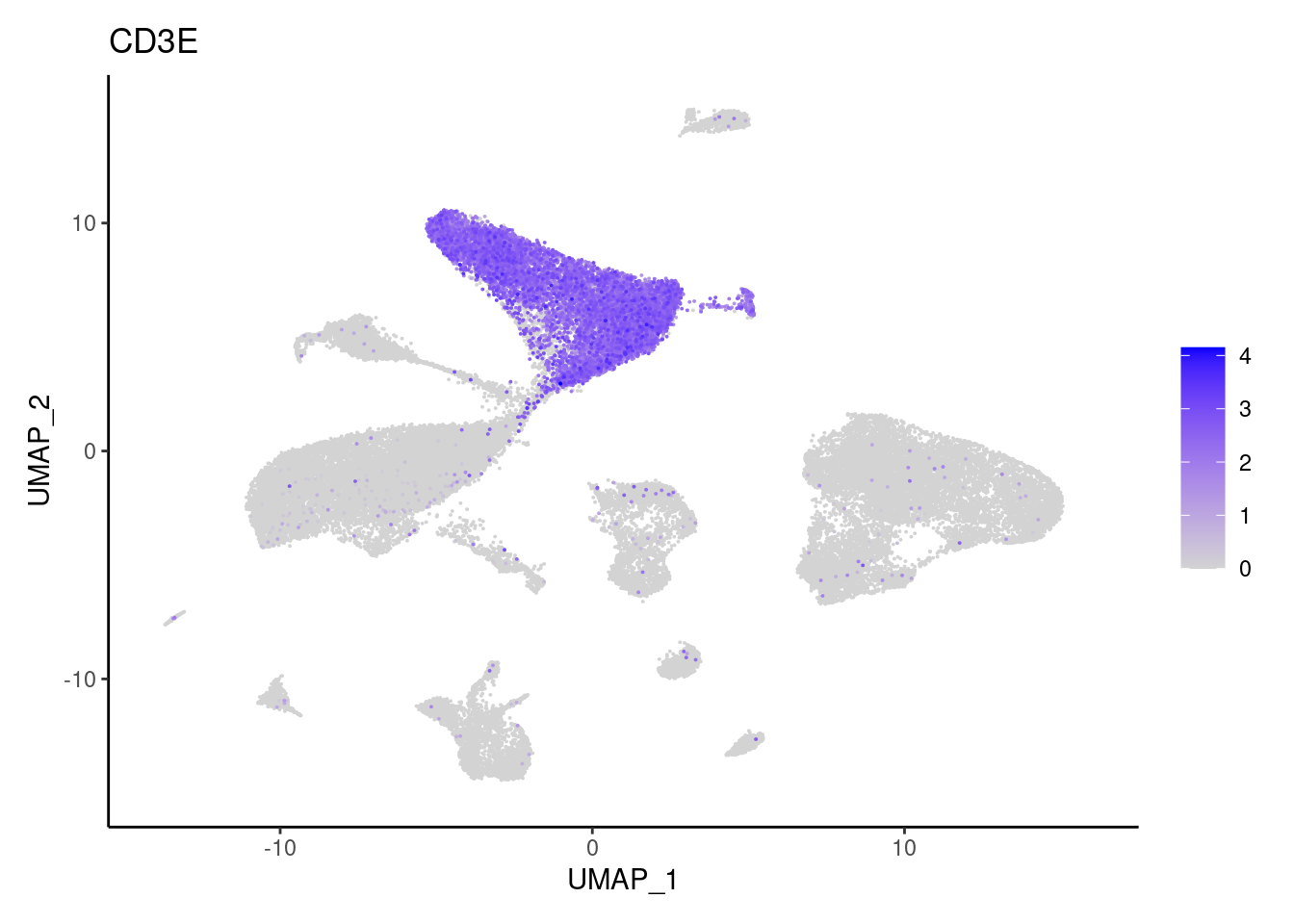
CD3G
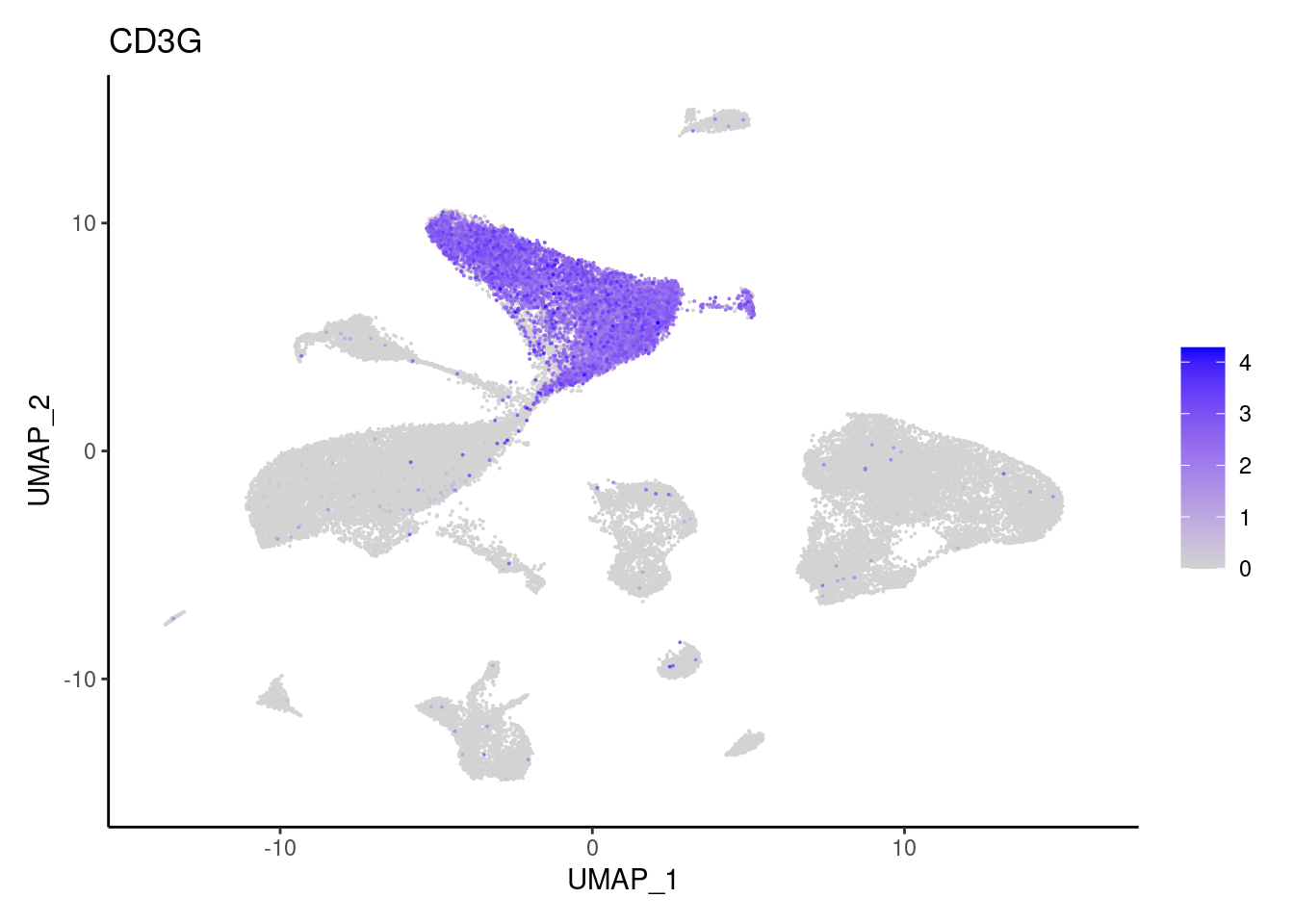
CD8A
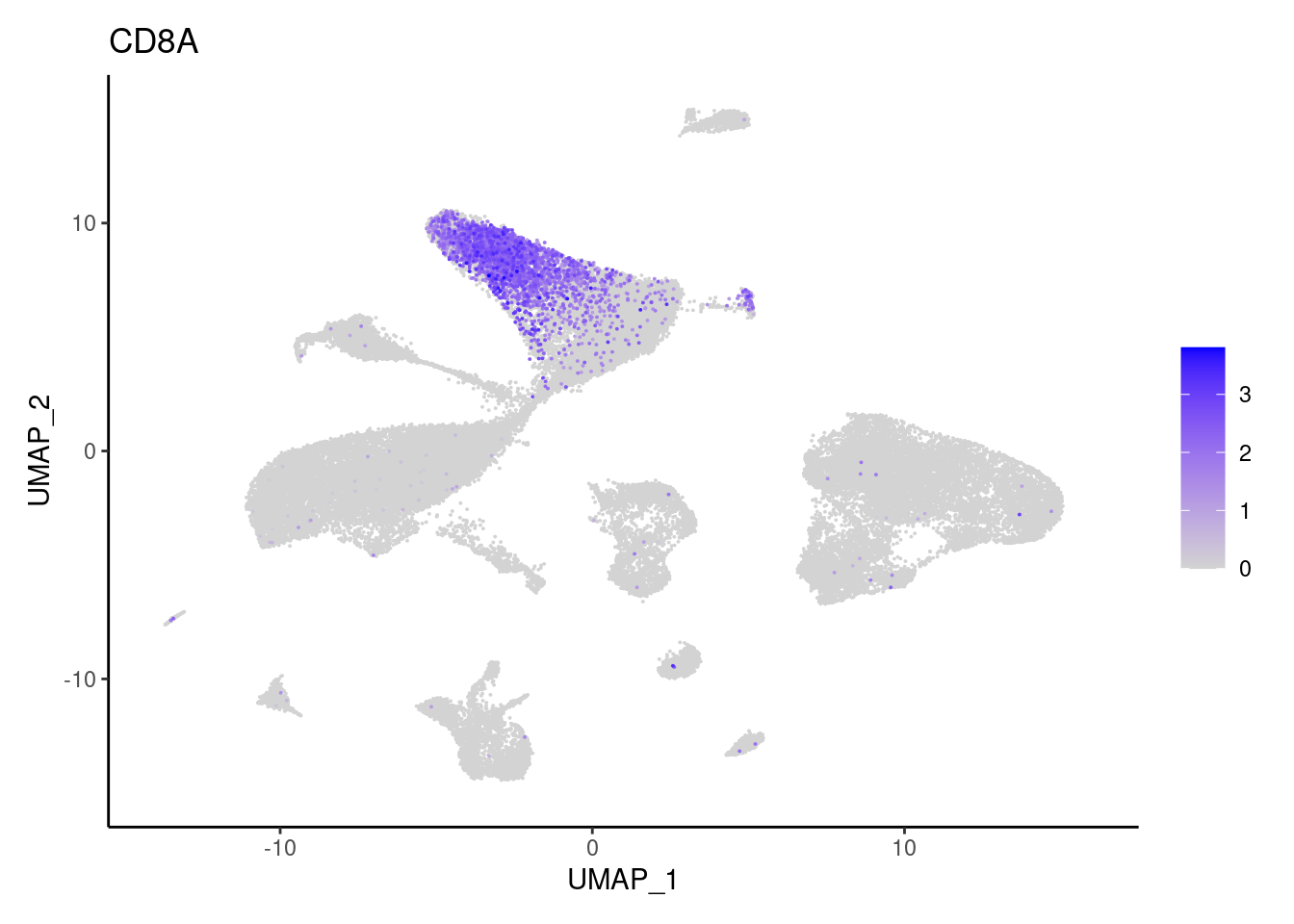
FOXP3
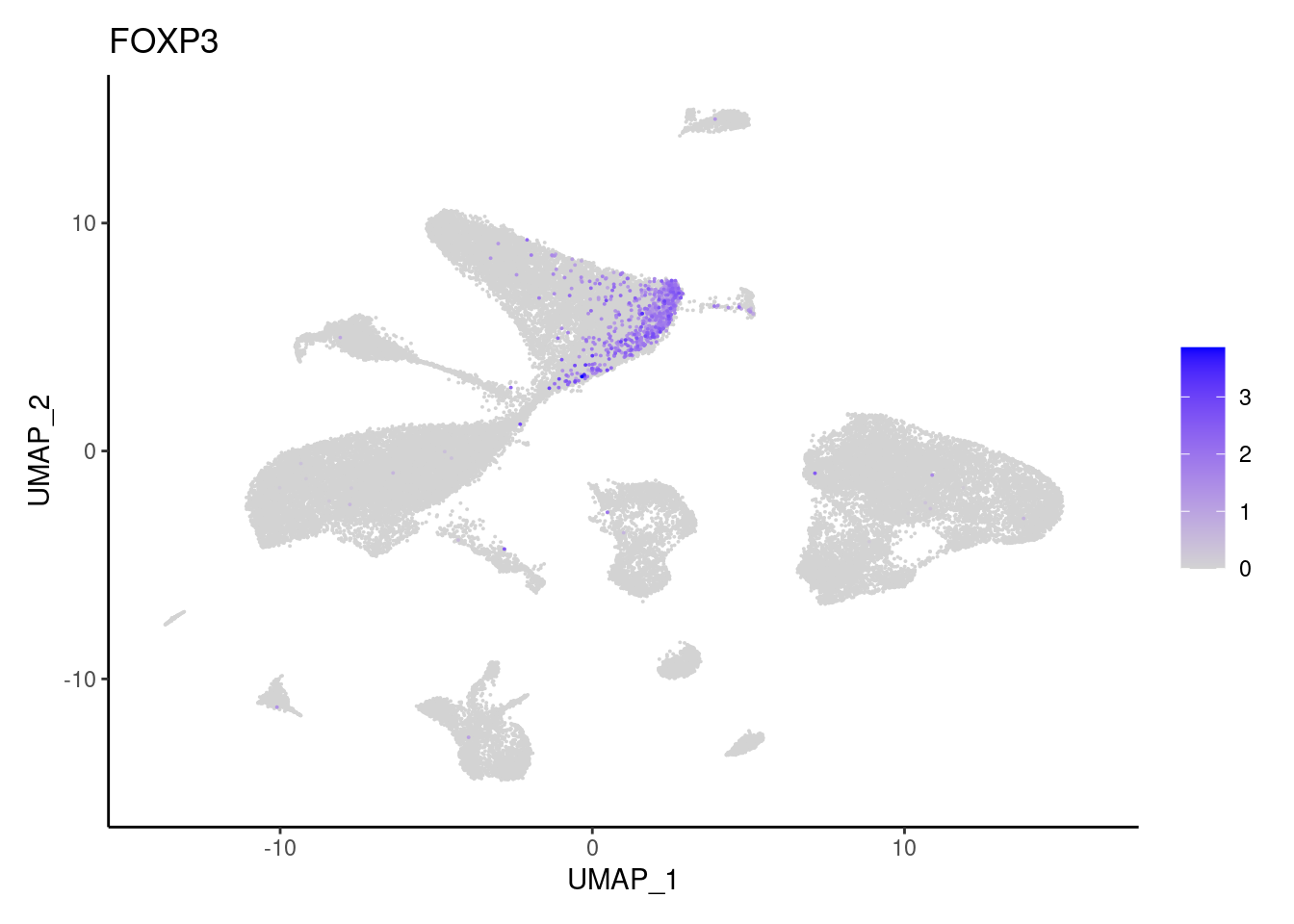
GZMA
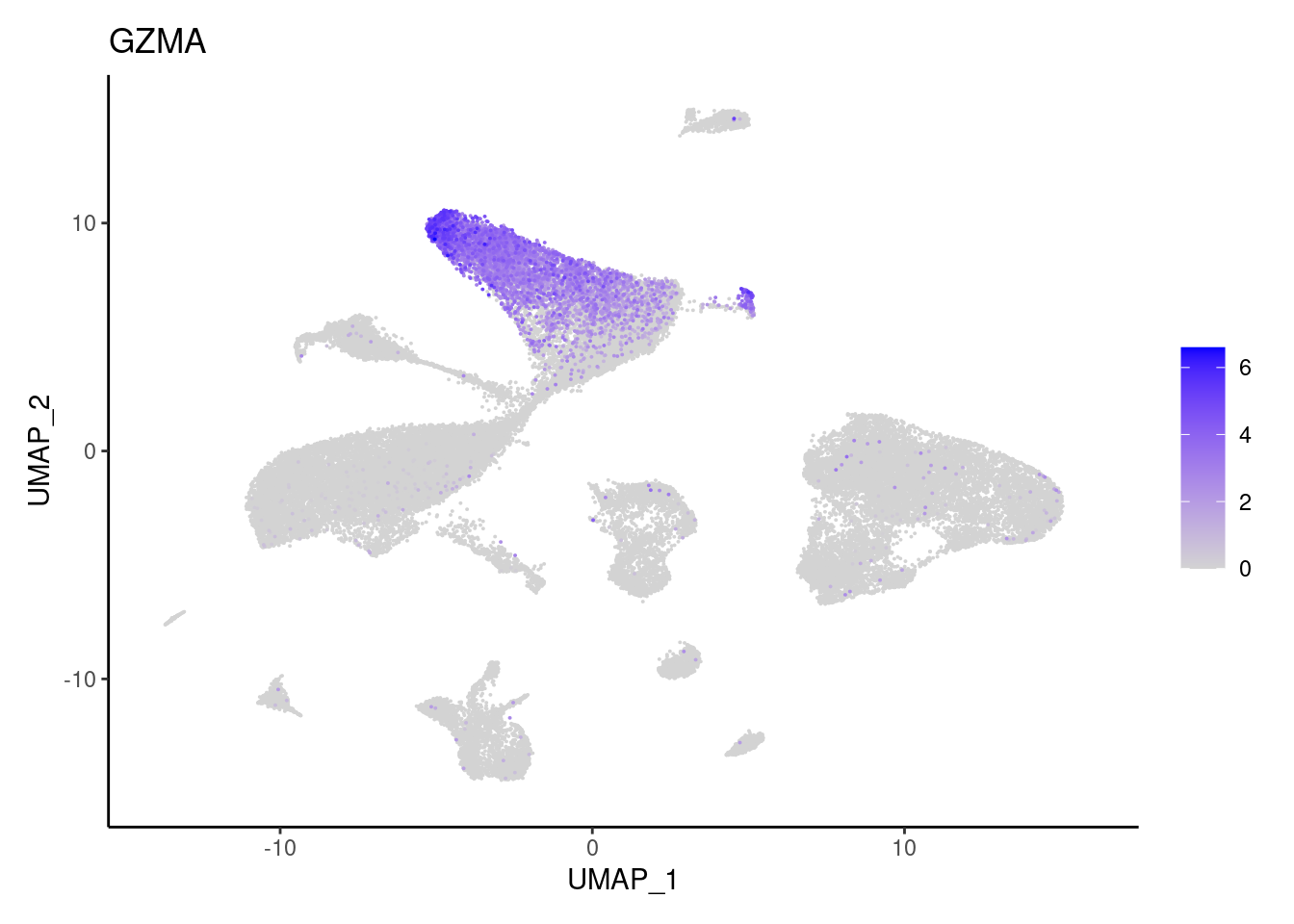
GZMB
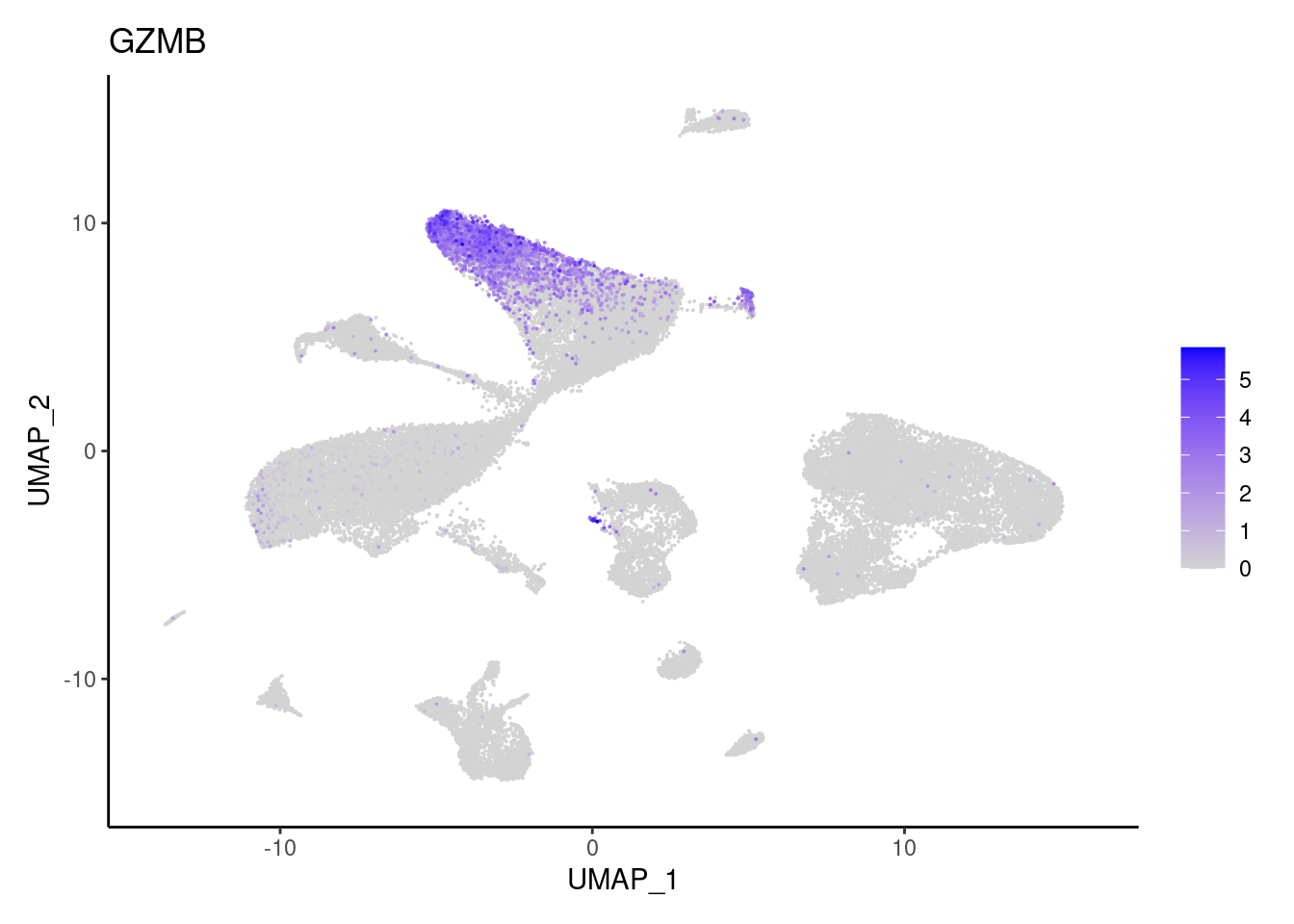
TRAC
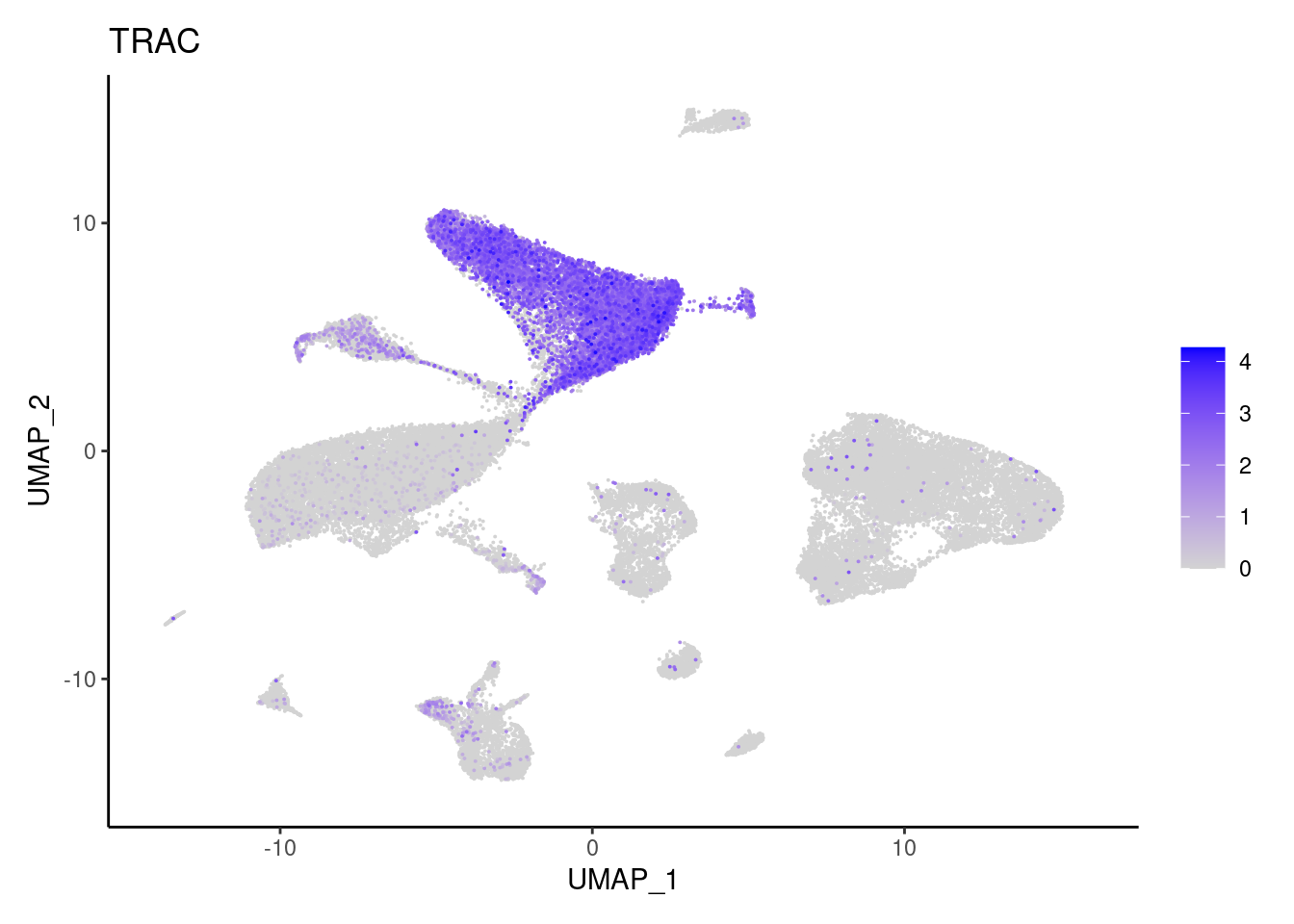
NKG7
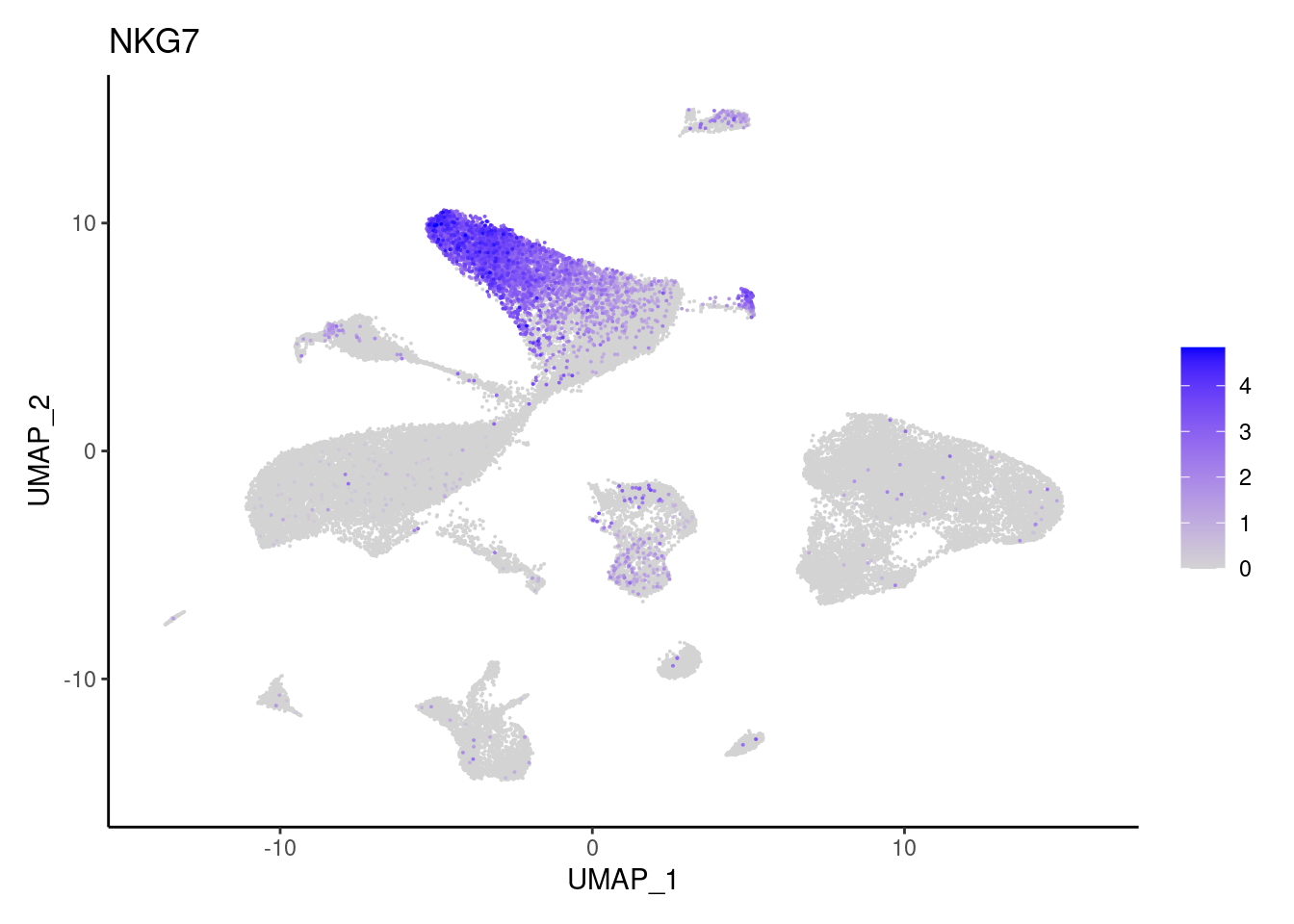
TRBC1
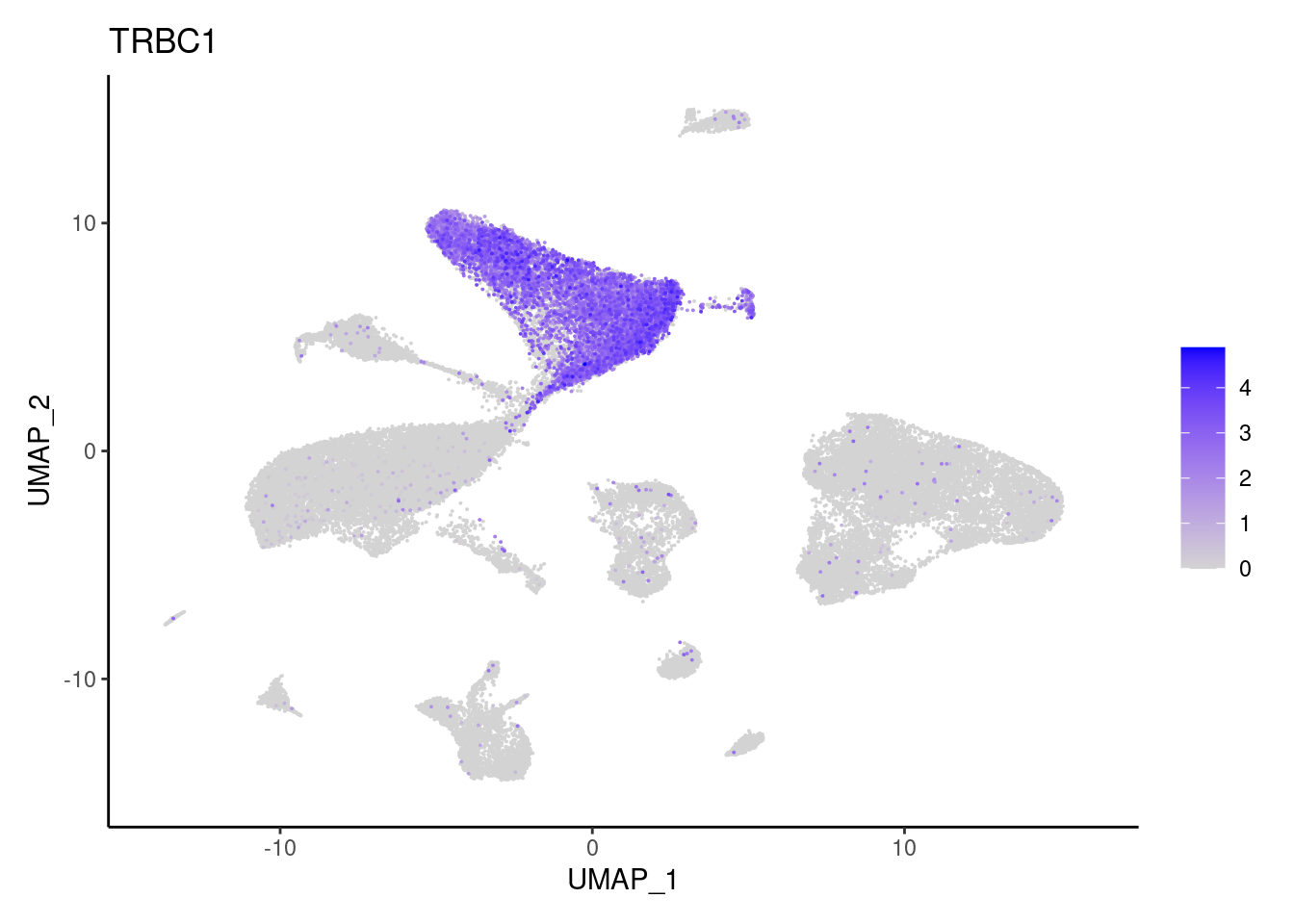
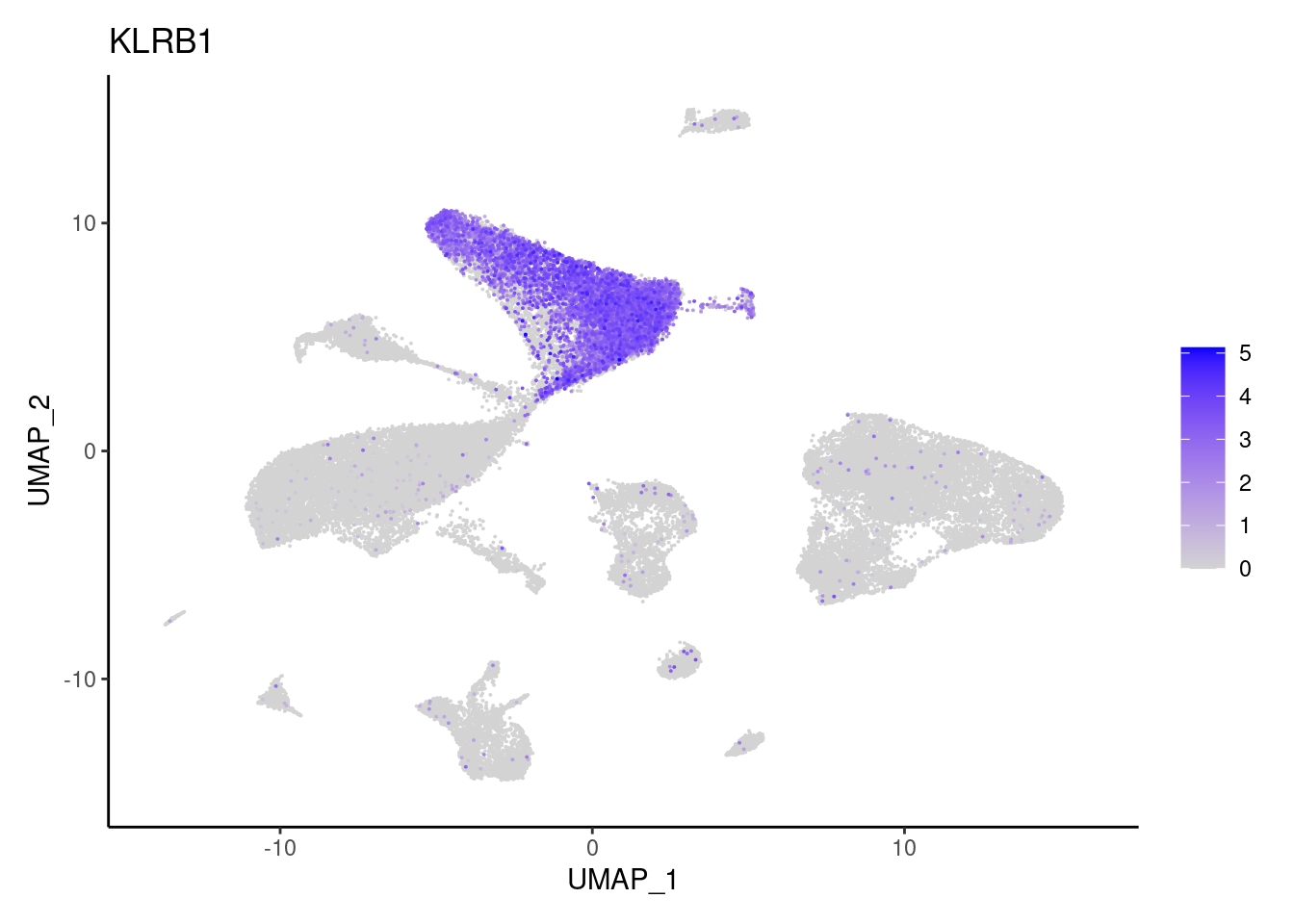
NK or NKT cells
KLRB1
cat("## Dotplot \n")Dotplot
DotPlot(seudataf, features = unique(subset_genes$gene), group.by = 'RNA_snn_res.0.1' ) + theme_classic() + theme(axis.text.x = element_text(angle=90, vjust = 0.5), axis.title = element_blank()) + NoLegend() + labs(title = 'Resolution 0.1')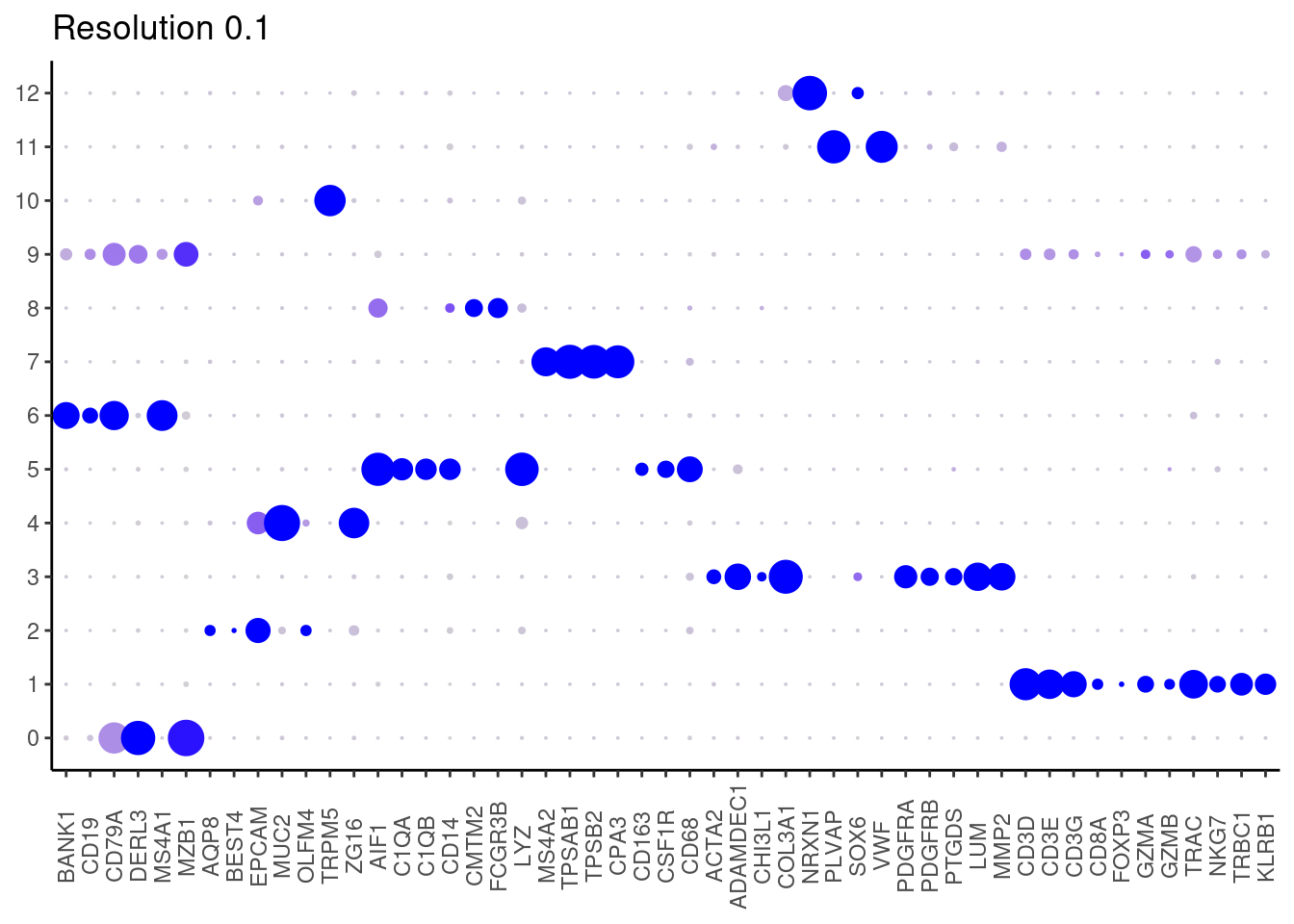
Subsets
We separate an extra subset of cycling cells and save each subset as a separated object.
myeloids <- seudataf[,seudataf$RNA_snn_res.0.1 %in% c(5,7,8)]
tcells <- seudataf[,seudataf$RNA_snn_res.0.1 %in% c(1)]
plasmas <- seudataf[,seudataf$RNA_snn_res.0.1 %in% c(0, 6)]
epi <- seudataf[,seudataf$RNA_snn_res.0.1 %in% c(2,4,10)]
stroma <- seudataf[,seudataf$RNA_snn_res.0.1 %in% c(12,11,3)]
cycling <- seudataf[,seudataf$RNA_snn_res.0.1 %in% c(9)]
dir.create('~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/SUBSETS/')
setwd('~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/SUBSETS/')
dir.create('FROM_SAMPLES_TOGETHER')
# save each subset as a separated object
saveRDS(myeloids,'FROM_SAMPLES_TOGETHER/myeloids.RDS')
saveRDS(tcells,'FROM_SAMPLES_TOGETHER/tcells.RDS')
saveRDS(plasmas,'FROM_SAMPLES_TOGETHER/plasmas.RDS')
saveRDS(epi,'FROM_SAMPLES_TOGETHER/epi.RDS')
saveRDS(stroma,'FROM_SAMPLES_TOGETHER/stroma.RDS')
saveRDS(cycling,'FROM_SAMPLES_TOGETHER/cycling.RDS')sessionInfo()
sessionInfo()## R version 4.1.2 (2021-11-01)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.5 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
## LAPACK: /opt/R/4.1.2/lib/R/lib/libRlapack.so
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8 LC_COLLATE=C.UTF-8
## [5] LC_MONETARY=C.UTF-8 LC_MESSAGES=C LC_PAPER=C.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] Matrix_1.4-0 nnet_7.3-17 matchSCore2_0.1.0 harmony_0.1.0
## [5] Rcpp_1.0.9 rmarkdown_2.18 pandoc_0.1.0 readxl_1.3.1
## [9] magick_2.7.3 data.table_1.14.2 BiocParallel_1.28.3 RColorBrewer_1.1-3
## [13] ggrepel_0.9.1 ggrastr_1.0.1 usethis_2.1.5 clustree_0.4.4
## [17] ggraph_2.0.5 readr_2.1.2 dplyr_1.0.10 cowplot_1.1.1
## [21] reshape_0.8.8 formulaic_0.0.8 patchwork_1.1.2 MASS_7.3-55
## [25] viridis_0.6.2 viridisLite_0.4.1 scDblFinder_1.8.0 scran_1.22.1
## [29] scater_1.22.0 scuttle_1.4.0 celda_1.10.0 beepr_1.3
## [33] DropletUtils_1.14.2 SingleCellExperiment_1.16.0 SummarizedExperiment_1.24.0 Biobase_2.54.0
## [37] GenomicRanges_1.46.1 GenomeInfoDb_1.30.1 IRanges_2.28.0 S4Vectors_0.32.4
## [41] BiocGenerics_0.40.0 MatrixGenerics_1.6.0 matrixStats_0.62.0 ggplot2_3.3.6
## [45] plyr_1.8.7 sp_1.5-0 SeuratObject_4.1.1 Seurat_4.1.0.9007
##
## loaded via a namespace (and not attached):
## [1] rsvd_1.0.5 ica_1.0-3 corpcor_1.6.10 assertive.properties_0.0-4
## [5] foreach_1.5.2 lmtest_0.9-40 rprojroot_2.0.3 crayon_1.5.2
## [9] spatstat.core_2.4-4 rhdf5filters_1.6.0 backports_1.4.1 nlme_3.1-155
## [13] rlang_1.0.6 XVector_0.34.0 ROCR_1.0-11 irlba_2.3.5
## [17] SparseM_1.81 limma_3.50.3 xgboost_1.5.0.2 rjson_0.2.21
## [21] bit64_4.0.5 glue_1.6.2 sctransform_0.3.4 parallel_4.1.2
## [25] vipor_0.4.5 spatstat.sparse_2.1-1 AnnotationDbi_1.56.2 spatstat.geom_2.4-0
## [29] tidyselect_1.1.2 fitdistrplus_1.1-8 tidyr_1.2.1 assertive.types_0.0-3
## [33] zoo_1.8-10 org.Mm.eg.db_3.14.0 xtable_1.8-4 magrittr_2.0.3
## [37] evaluate_0.18 cli_3.4.0 zlibbioc_1.40.0 rstudioapi_0.13
## [41] miniUI_0.1.1.1 bslib_0.4.1 rpart_4.1.16 RcppEigen_0.3.3.9.2
## [45] shiny_1.7.3 BiocSingular_1.10.0 xfun_0.34 clue_0.3-60
## [49] cluster_2.1.2 tidygraph_1.2.1 KEGGREST_1.34.0 tibble_3.1.8
## [53] listenv_0.8.0 Biostrings_2.62.0 png_0.1-7 future_1.28.0
## [57] withr_2.5.0 bitops_1.0-7 ggforce_0.3.3 cellranger_1.1.0
## [61] assertive.base_0.0-9 dqrng_0.3.0 pillar_1.8.1 GlobalOptions_0.1.2
## [65] cachem_1.0.6 fs_1.5.2 GetoptLong_1.0.5 DelayedMatrixStats_1.16.0
## [69] vctrs_0.4.1 ellipsis_0.3.2 generics_0.1.3 tools_4.1.2
## [73] beeswarm_0.4.0 munsell_0.5.0 tweenr_1.0.2 DelayedArray_0.20.0
## [77] fastmap_1.1.0 compiler_4.1.2 pkgload_1.2.4 abind_1.4-5
## [81] httpuv_1.6.6 plotly_4.10.0 rgeos_0.5-9 GenomeInfoDbData_1.2.7
## [85] gridExtra_2.3 enrichR_3.0 edgeR_3.36.0 lattice_0.20-45
## [89] deldir_1.0-6 utf8_1.2.2 later_1.3.0 jsonlite_1.8.3
## [93] multipanelfigure_2.1.2 scales_1.2.1 graph_1.72.0 ScaledMatrix_1.2.0
## [97] pbapply_1.5-0 sparseMatrixStats_1.6.0 lazyeval_0.2.2 promises_1.2.0.1
## [101] doParallel_1.0.17 R.utils_2.12.0 goftest_1.2-3 checkmate_2.0.0
## [105] spatstat.utils_2.3-1 reticulate_1.26 textshaping_0.3.6 statmod_1.4.36
## [109] Rtsne_0.16 uwot_0.1.14 igraph_1.3.4 HDF5Array_1.22.1
## [113] survival_3.2-13 rsconnect_0.8.25 yaml_2.3.6 systemfonts_1.0.4
## [117] htmltools_0.5.3 memoise_2.0.1 locfit_1.5-9.6 graphlayouts_0.8.0
## [121] digest_0.6.30 assertthat_0.2.1 rappdirs_0.3.3 mime_0.12
## [125] RSQLite_2.2.17 future.apply_1.9.1 blob_1.2.3 R.oo_1.25.0
## [129] ragg_1.2.1 splines_4.1.2 labeling_0.4.2 Rhdf5lib_1.16.0
## [133] RCurl_1.98-1.8 assertive.numbers_0.0-2 hms_1.1.1 rhdf5_2.38.1
## [137] colorspace_2.0-3 ggbeeswarm_0.6.0 shape_1.4.6 assertive.files_0.0-2
## [141] sass_0.4.2 RANN_2.6.1 circlize_0.4.14 audio_0.1-10
## [145] fansi_1.0.3 tzdb_0.2.0 brio_1.1.3 parallelly_1.32.1
## [149] R6_2.5.1 grid_4.1.2 ggridges_0.5.3 lifecycle_1.0.3
## [153] formatR_1.12 bluster_1.4.0 curl_4.3.2 jquerylib_0.1.4
## [157] leiden_0.4.3 testthat_3.1.2 desc_1.4.0 RcppAnnoy_0.0.19
## [161] org.Hs.eg.db_3.14.0 iterators_1.0.14 stringr_1.4.1 topGO_2.46.0
## [165] htmlwidgets_1.5.4 beachmat_2.10.0 polyclip_1.10-0 purrr_0.3.4
## [169] gridGraphics_0.5-1 ComplexHeatmap_2.10.0 mgcv_1.8-38 globals_0.16.1
## [173] spatstat.random_2.2-0 progressr_0.11.0 codetools_0.2-18 GO.db_3.14.0
## [177] metapod_1.2.0 MCMCprecision_0.4.0 R.methodsS3_1.8.2 gtable_0.3.1
## [181] DBI_1.1.3 highr_0.9 tensor_1.5 httr_1.4.4
## [185] KernSmooth_2.23-20 vroom_1.5.7 stringi_1.7.8 reshape2_1.4.4
## [189] farver_2.1.1 combinat_0.0-8 BiocNeighbors_1.12.0 scattermore_0.8
## [193] bit_4.0.4 spatstat.data_2.2-0 pkgconfig_2.0.3 corrplot_0.92
## [197] knitr_1.40