B and plasmacells subset analysis
Last compiled on 17 November, 2022
We considered five major cell subsets: (1) myeloid, (2) T, (3) B and plasma, (4) stromal, and (5) epithelial cells. We classified cells into these subsets according to their gene expression. We consider in this file the cells classified as B and Plasmacells. We reanalyze and QC the data, and look for the refined clusters.
Load libraries
library(Seurat)
library(plyr)
library(dplyr)
library(ggplot2)
library(DropletUtils)
library(celda)
library(SingleCellExperiment)
library(scater)
library(scran)
library(scDblFinder)
library(viridis)
library(MASS)
library(patchwork)
library(readr)Load extra sources
get_density <- function(x, y, ...) { # function from https://slowkow.com/notes/ggplot2-color-by-density/
dens <- MASS::kde2d(x, y, ...)
ix <- findInterval(x, dens$x)
iy <- findInterval(y, dens$y)
ii <- cbind(ix, iy)
return(dens$z[ii])
}
fancy_scientific <- function(l) { # function from https://stackoverflow.com/a/24241954
# turn in to character string in scientific notation
l <- format(l, scientific = TRUE)
# quote the part before the exponent to keep all the digits
l <- gsub("^(.*)e", "'\\1'e", l)
# turn the 'e+' into plotmath format
l <- gsub("e", "%*%10^", l)
# return this as an expression
parse(text=l)
}
setwd("~/000_GitHub/ibd-bcn_single_cell")
source('source/functions_scrnaseq.R')
source('source/colors.R')Load the data
setwd('~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/')
plasmas <- readRDS('SUBSETS/FROM_SAMPLES_TOGETHER/plasmas.RDS')
cycling <- readRDS('SUBSETS/FROM_SAMPLES_TOGETHER/cycling.RDS')
plasmas <- merge(plasmas, cycling)Reanalysis
These are the clusters we selected as B cells, Plasmacells, and cycling cells in the “02_Data_to_subsets” file. Cycling cells were a compendium of T, B, plasma cells. We will remove the cells not belonging to this subset during the QC.
QC
MT-gene expression
Distribution of cells in scatter plot (nfeatures/percent.mt) to check where are the majority of our cells. We can see the dots colored by density using the function get_density shown in [Load extra functions and sources].
meta <- plasmas@meta.data
meta$density <- get_density(meta$percent.mt, meta$nFeature_RNA, n = 1000)
ggplot(meta) +
geom_point(aes(percent.mt, nFeature_RNA, color = density), size = 0.2) +
scale_color_viridis() + theme_classic() +
scale_y_sqrt()+ scale_x_sqrt(breaks = c(1,10,100), limits = c(0,100))+
geom_vline(xintercept = 25, linetype = 2, color = 'darkgray')+
theme(text = element_text( size = 12),
axis.title = element_text( size = 12),
legend.text = element_blank())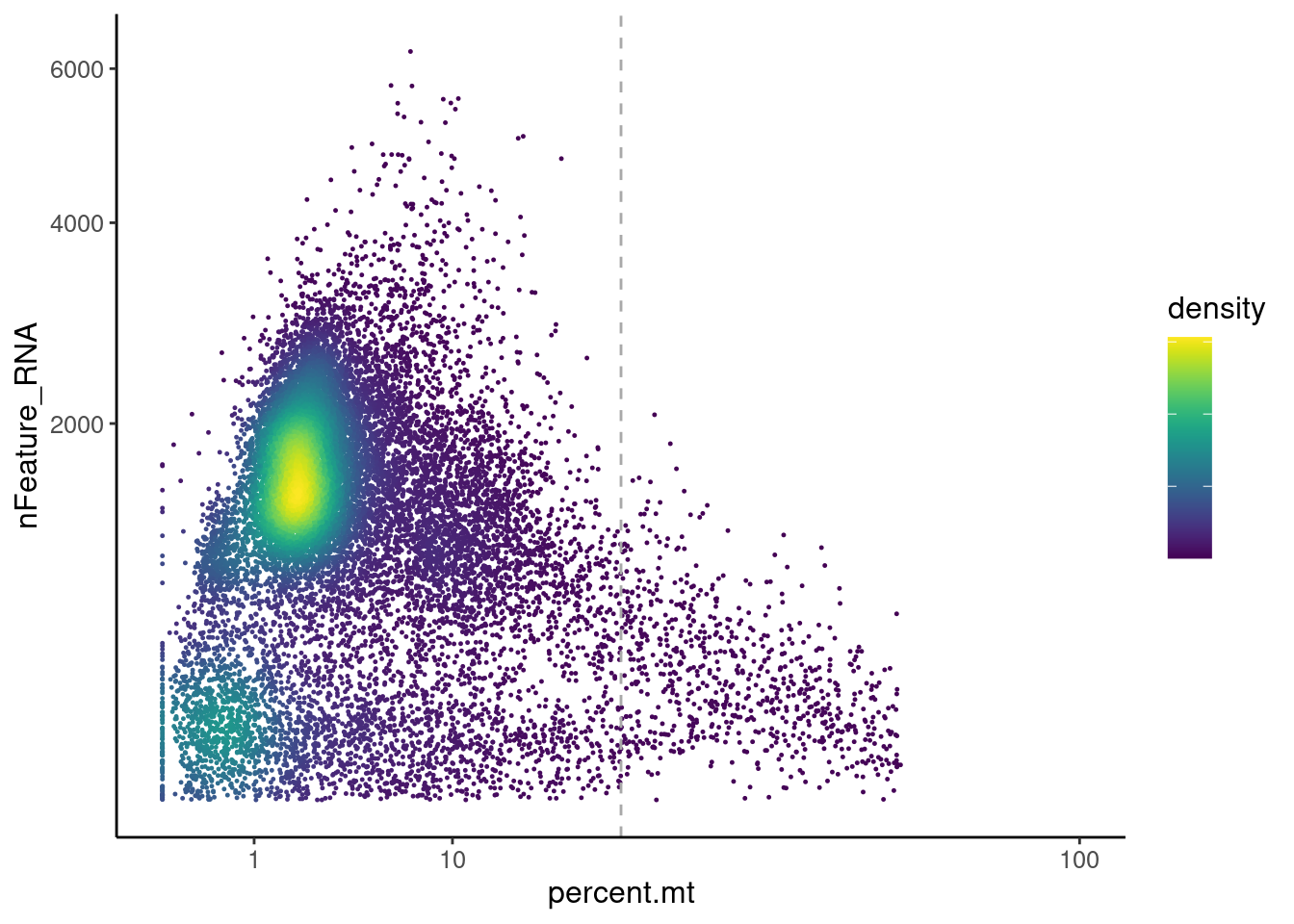 We analyzed the data using different cutoffs for the percent.mt (data
not shown) and finally decided that 25% was a reasonable choice for our
dataset.
We analyzed the data using different cutoffs for the percent.mt (data
not shown) and finally decided that 25% was a reasonable choice for our
dataset.
plasmas <- plasmas[,plasmas$percent.mt < 25]Non-B or Plasmacell genes
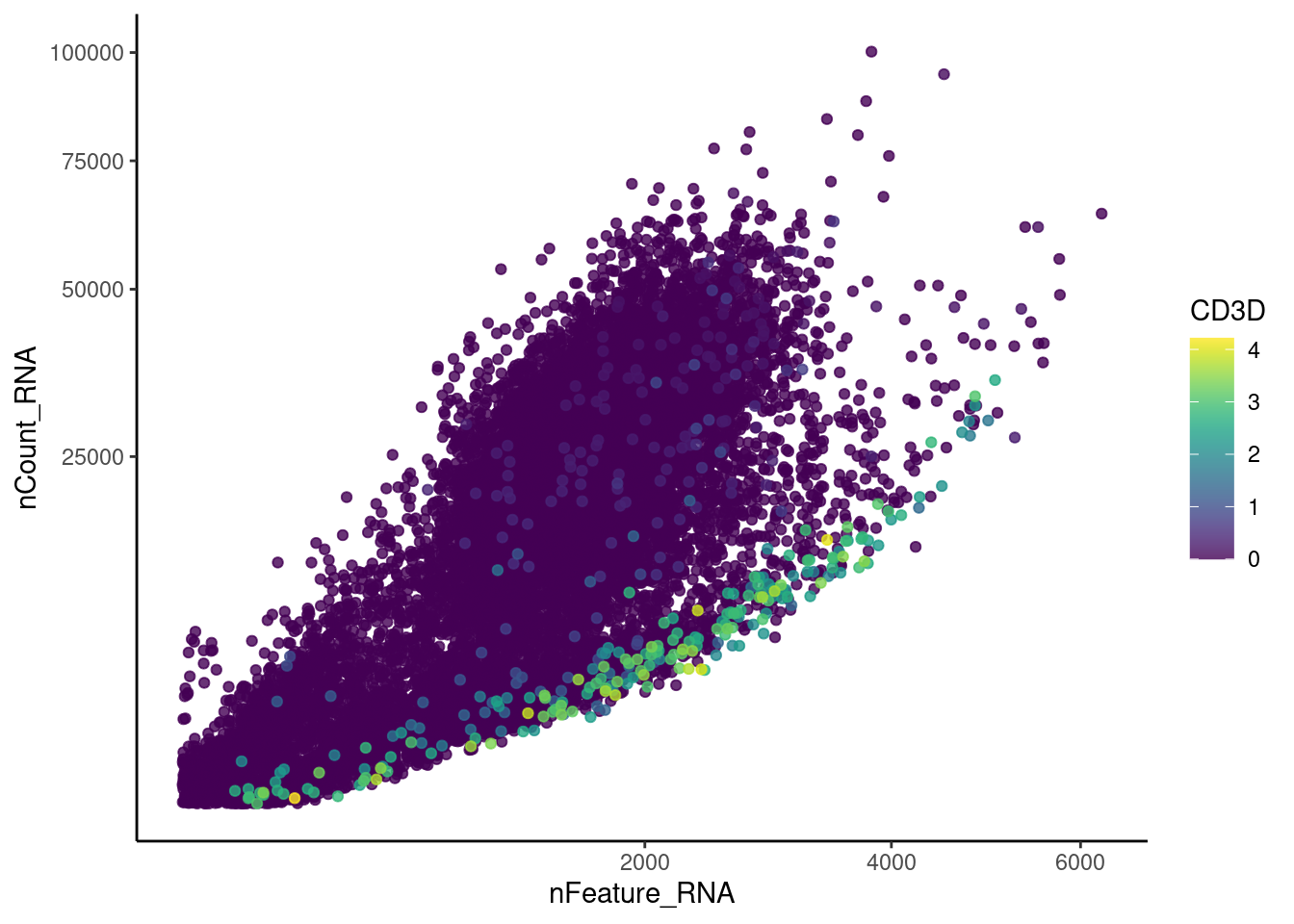
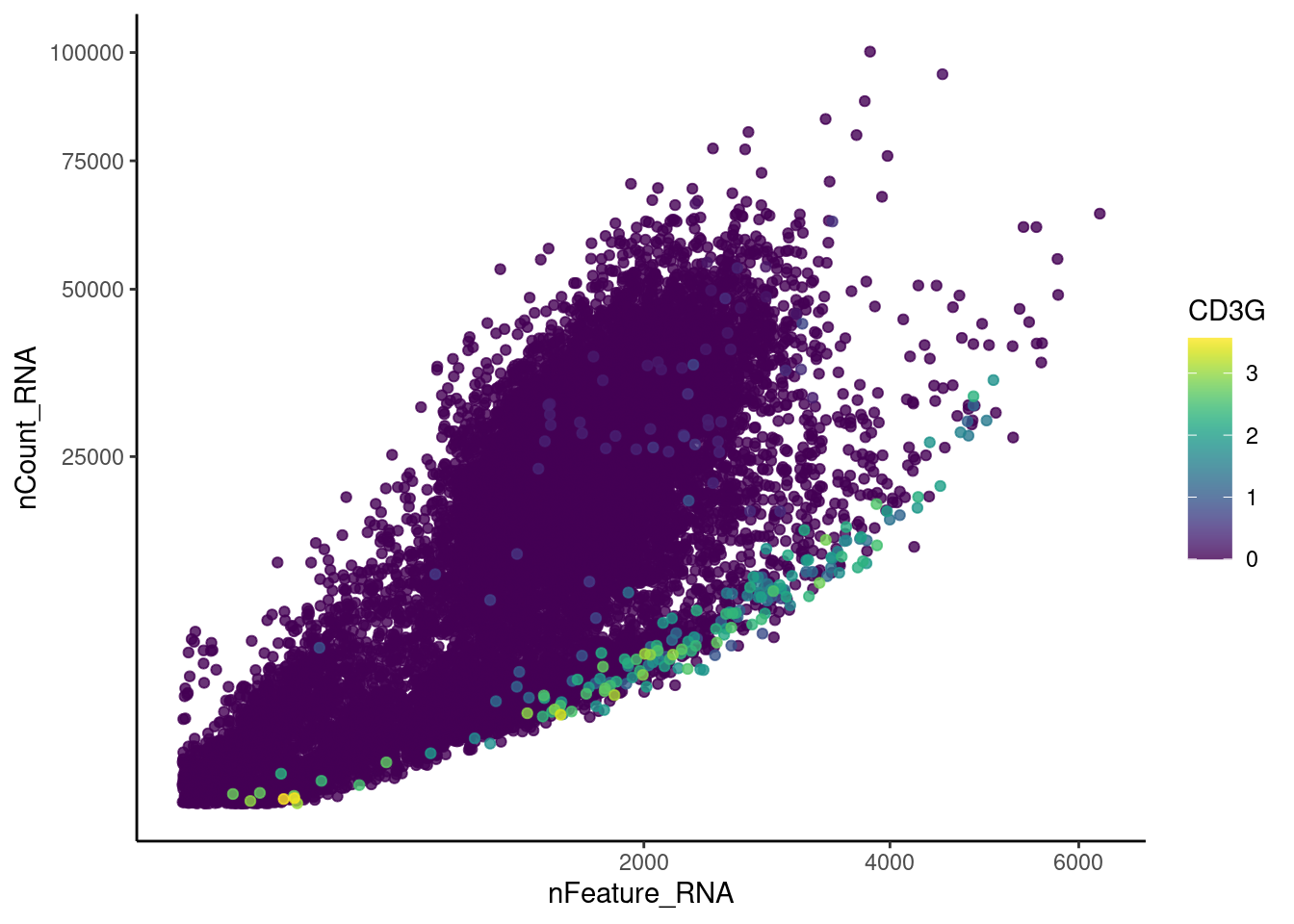
Right now plasmas has 28287 features across 15373 cells.
Nevertheless, many cells express genes that we know should not be
expressed by B or plasmacells. We will remove those cells.
genes <- c('CD3E','CD3D','CD3G','C1QA','EPCAM')
for (gene in genes) {
jd <- FetchData(plasmas, vars = c('nCount_RNA', 'nFeature_RNA', gene))
jd <- jd[order(jd[,ncol(jd)]),]
k <- ggplot(jd, mapping = aes_string(x = 'nFeature_RNA', y = 'nCount_RNA', color = gene))+
geom_point() + theme_classic() + scale_color_viridis(alpha = 0.8) + scale_x_sqrt() + scale_y_sqrt()
cat("#### ", gene, "\n"); print(k); cat("\n\n")
}CD3E
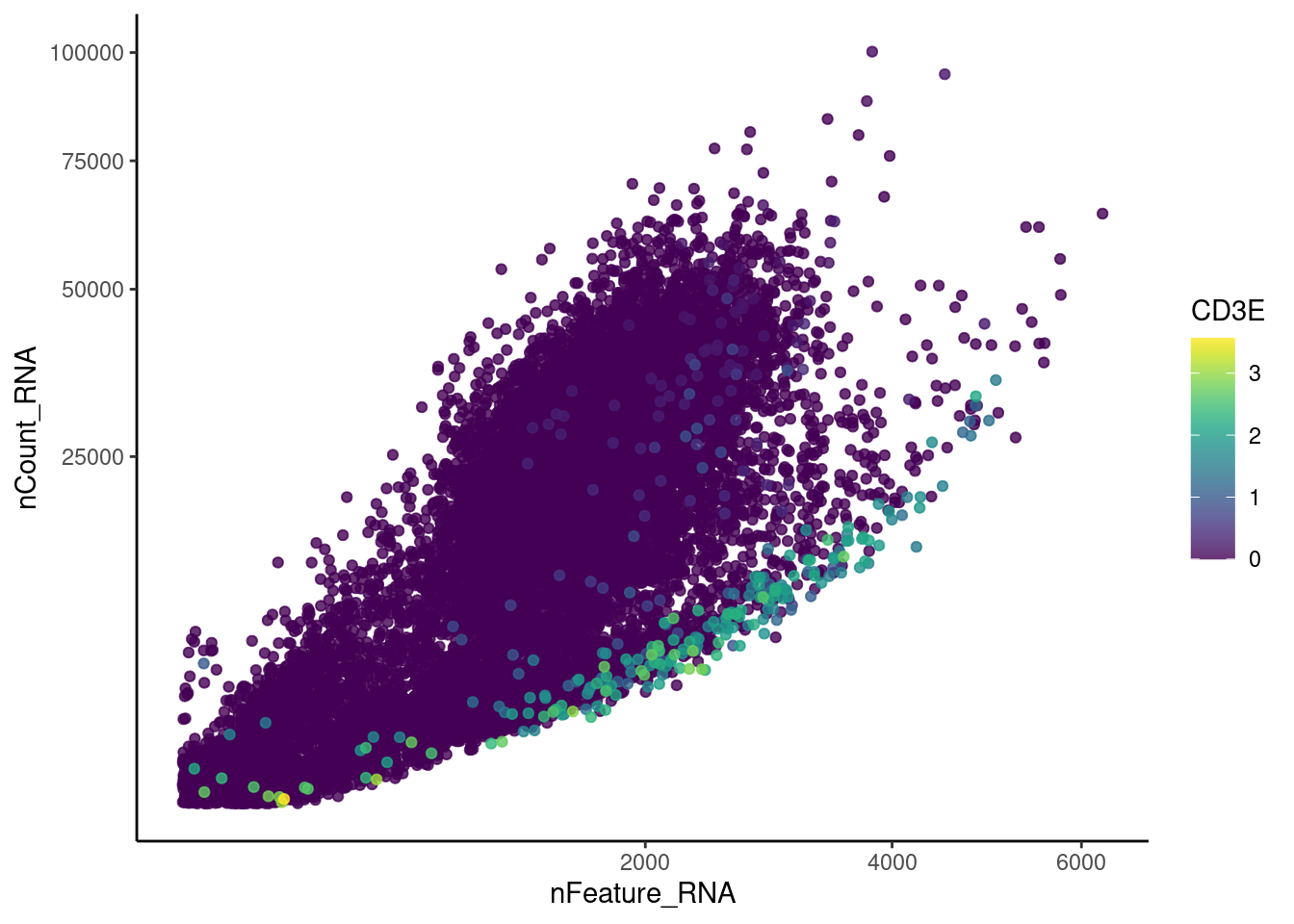
CD3D
CD3G
C1QA
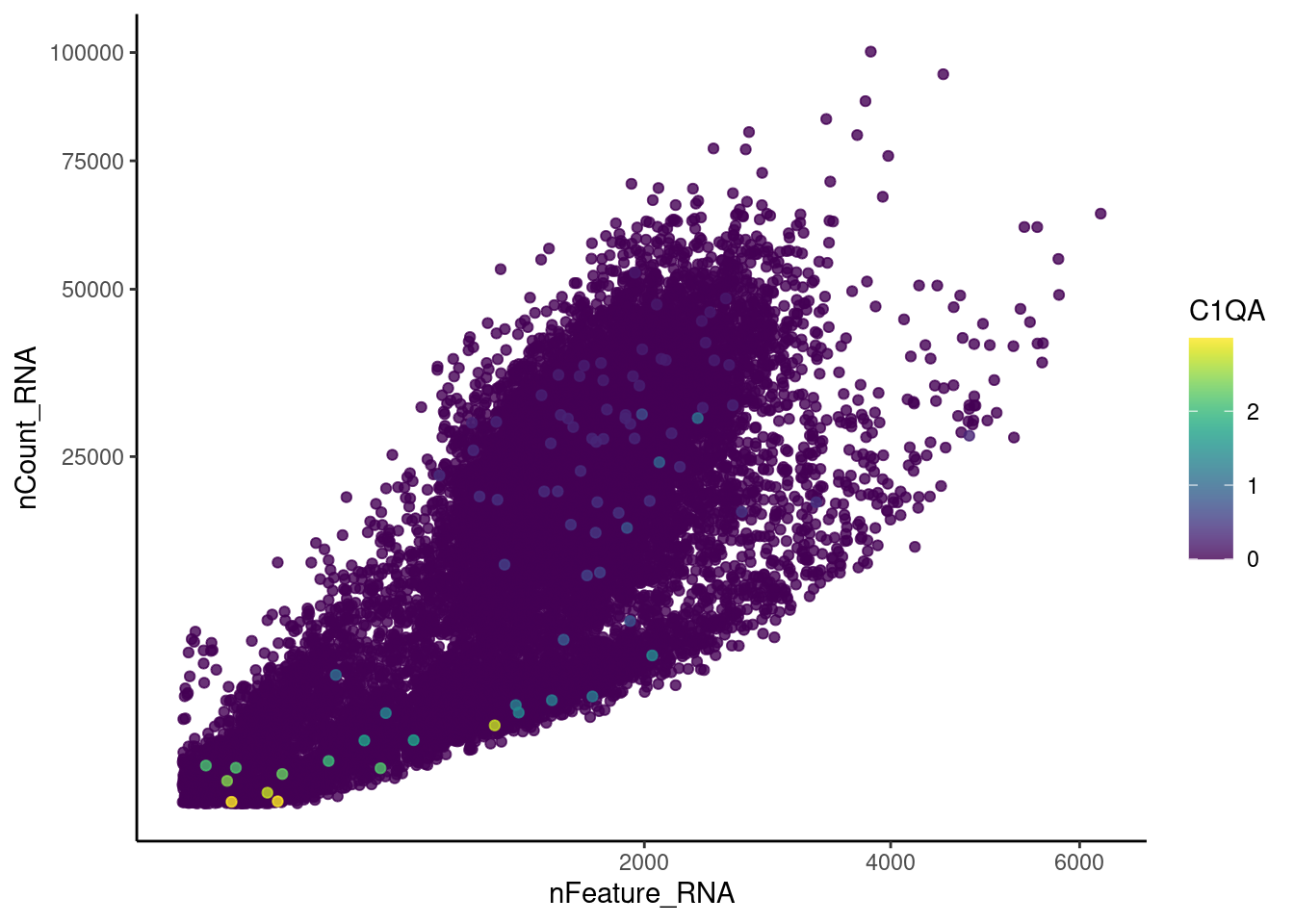
EPCAM
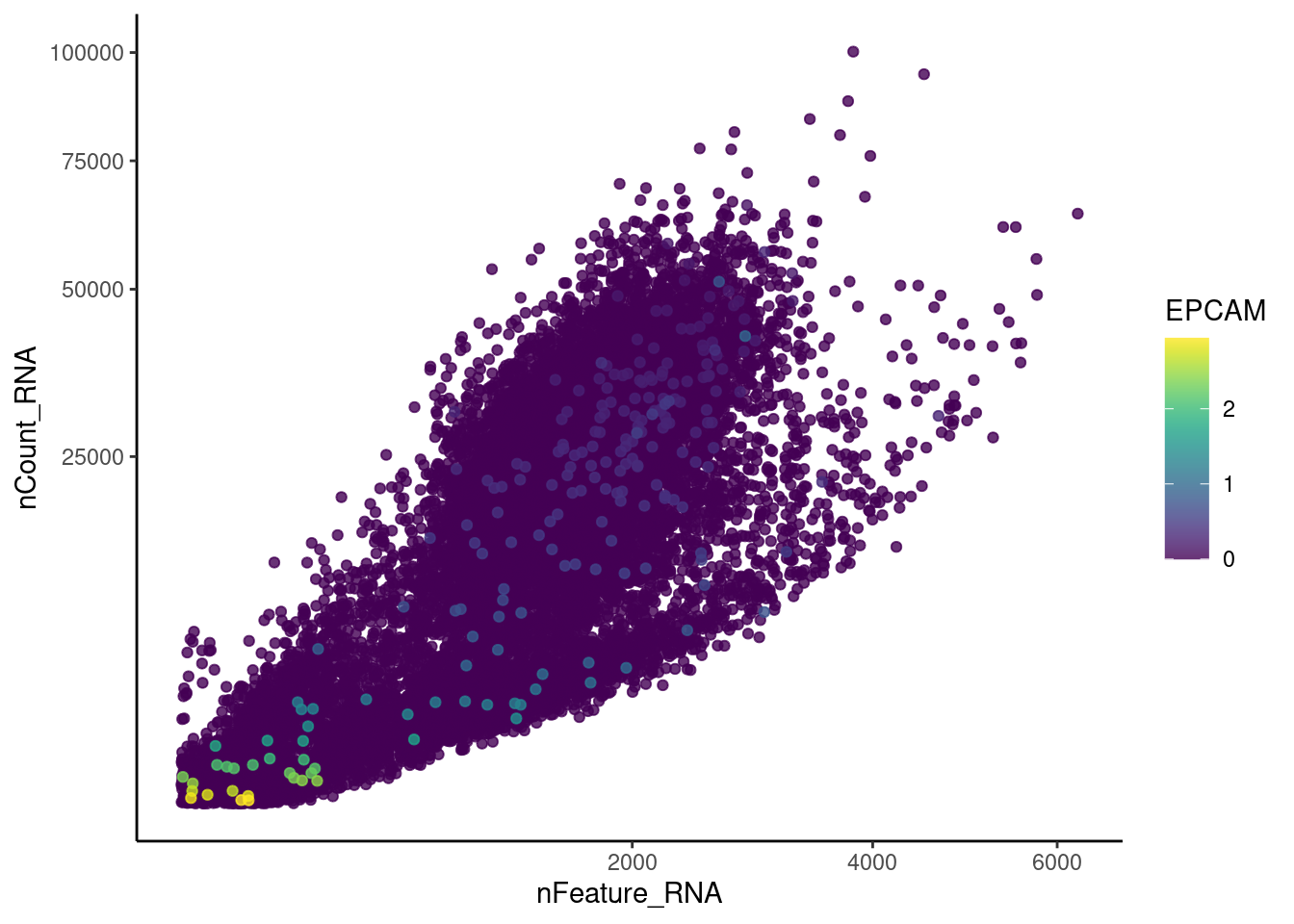
cat("#### code removal \n")code removal
counts <- plasmas@assays$RNA@counts
p <- grep("CD3E$|CD3D$|CD3G$|^C1QA$|^EPCAM$",rownames(plasmas))
pp <- which(Matrix::colSums(counts[p,])>0)
xx <-setdiff(colnames(plasmas), names(pp))
plasmas <- subset(plasmas,cells = xx)
cat('\n\n')Genes wo expression
We remove the genes without expression from the dataset.
counts <- plasmas@assays$RNA@counts
pp <- which(Matrix::rowSums(counts)==0)
xx <-setdiff(rownames(plasmas), names(pp))
plasmas <- subset(plasmas, features = xx)Dimension reduction
plasmas <- seurat_to_pca(plasmas)
a <- ElbowPlot(plasmas, ndims = 100) +
geom_vline(xintercept = 25, colour="#BB0000", linetype = 2)+
labs(title = paste0('Elbowplot')) + theme_classic()
plasmas<- FindNeighbors(plasmas, dims = 1:25)
plasmas<-RunUMAP(plasmas, dims=1:25)
b <- DimPlot(plasmas, group.by = 'sample_name') +
labs(title = '25 PCS') +
theme_classic() +
theme(legend.position = 'bottom')
(a+b) / guide_area() +
plot_layout(heights = c(0.7,0.3),guides = 'collect')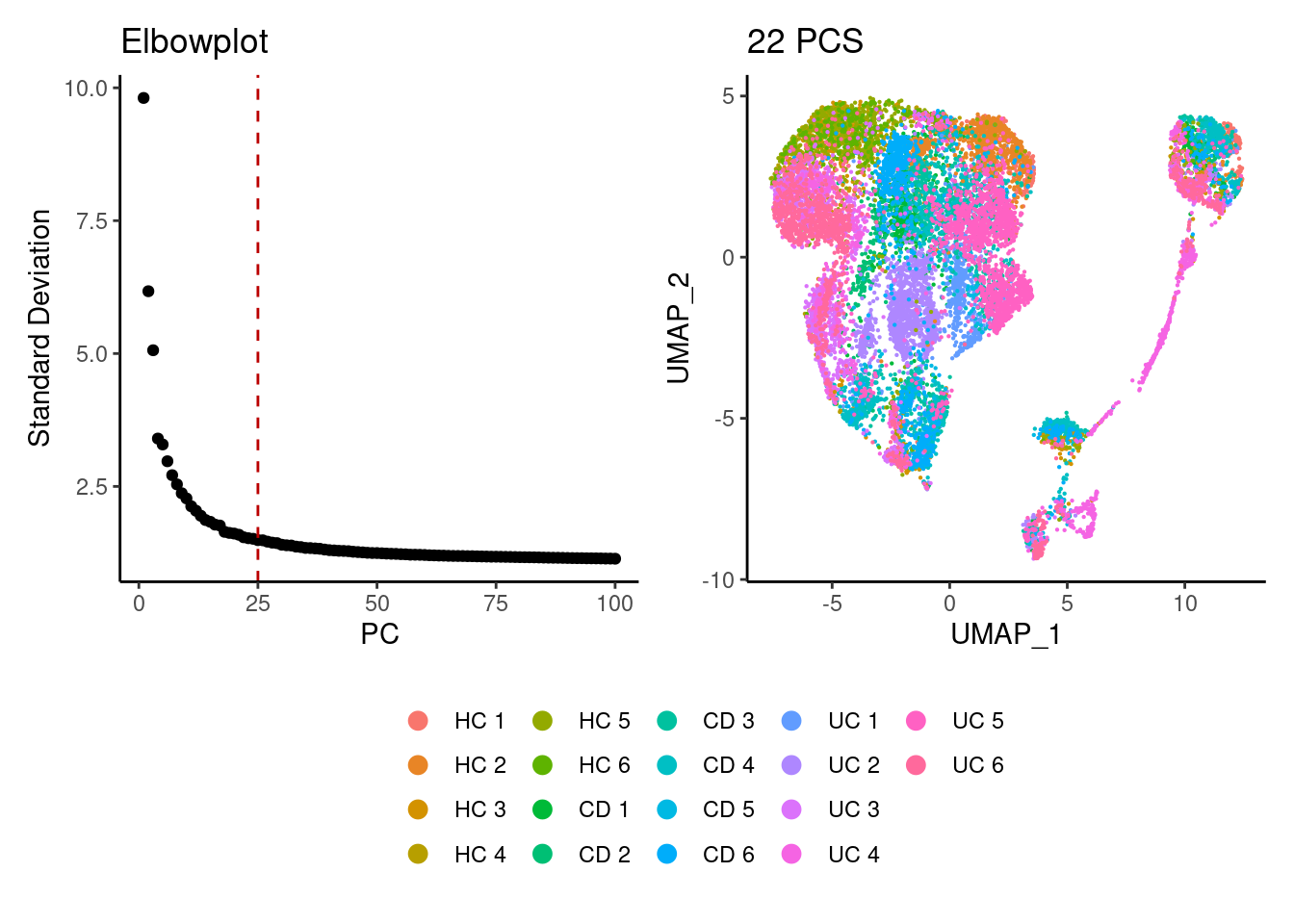
Harmony correction
plasmas <- RunHarmony(plasmas, group.by = 'sample', dims.use = 1:25)
a <- ElbowPlot(plasmas, ndims = 100) +
geom_vline(xintercept = 18, colour="#BB0000", linetype = 2)+
labs(title = paste0('Elbowplot'))+ theme_classic()
plasmas<- FindNeighbors(plasmas, reduction = "harmony", dims = 1:18)
plasmas<-RunUMAP(plasmas, dims=1:18, reduction= "harmony")
b <- DimPlot(plasmas, group.by = 'sample_name', pt.size = 0.1) +
theme_classic() +
theme(legend.position = 'bottom')
(a+b) / guide_area() +
plot_layout(heights = c(0.7,0.3),guides = 'collect')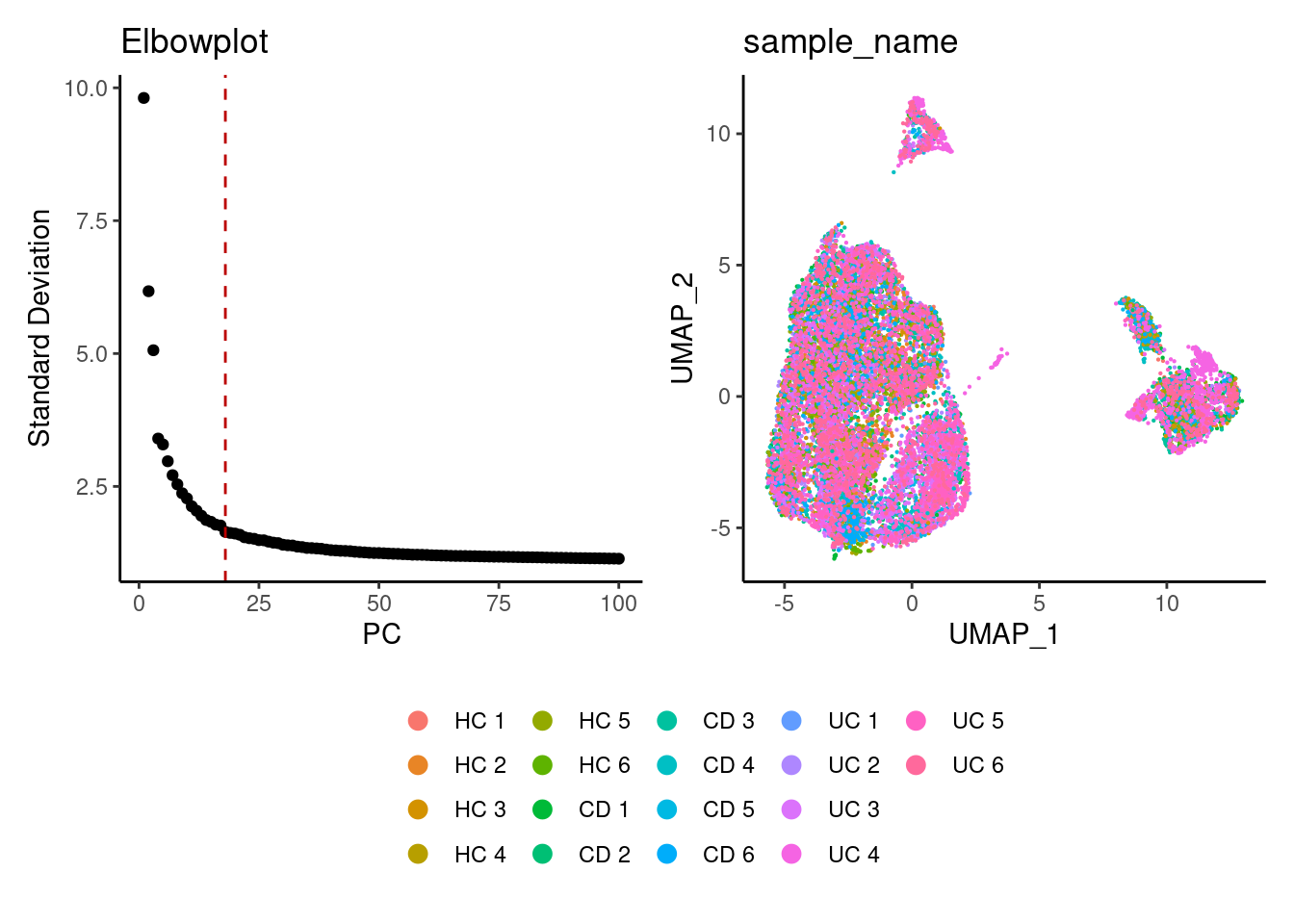
Louvain Clusterization
# dir.create('Markers')
# dir.create('Markers/BandPlasmacells')
path <- '~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/SUBSETS/Markers'
setwd(path)
plasmas <- resolutions(plasmas,
workingdir = path,
title = 'BandPlasmacells/Markers_plasmas_')
path <- '~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/SUBSETS/ON_THEIR_OWN/'
setwd(path)
saveRDS(plasmas, file = paste0(path,'plasmas_filtered.RDS'))Dimplot Resolutions
path <- '~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/SUBSETS/ON_THEIR_OWN/'
plasmas <- readRDS(file = paste0(path,'plasmas_filtered.RDS'))
for(i in c('0.1','0.3','0.5','0.7','0.9','1.1','1.3','1.5')){
j <-DimPlot(plasmas, group.by = paste0('RNA_snn_res.',i), label=T) +
theme_classic() +
theme(legend.position = 'none')
cat("##### ", i, "\n"); print(j); cat("\n\n")
}0.1
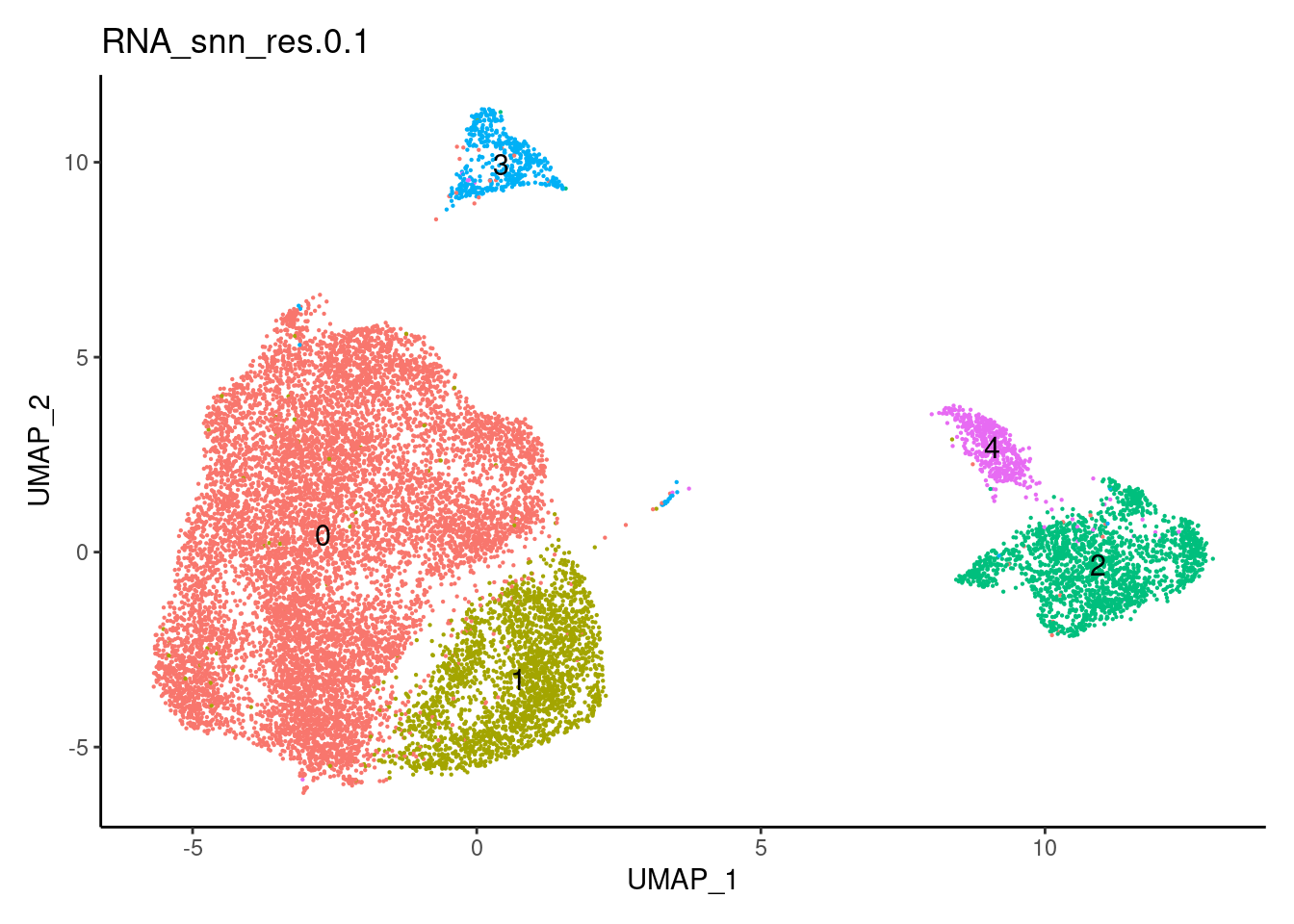
0.3
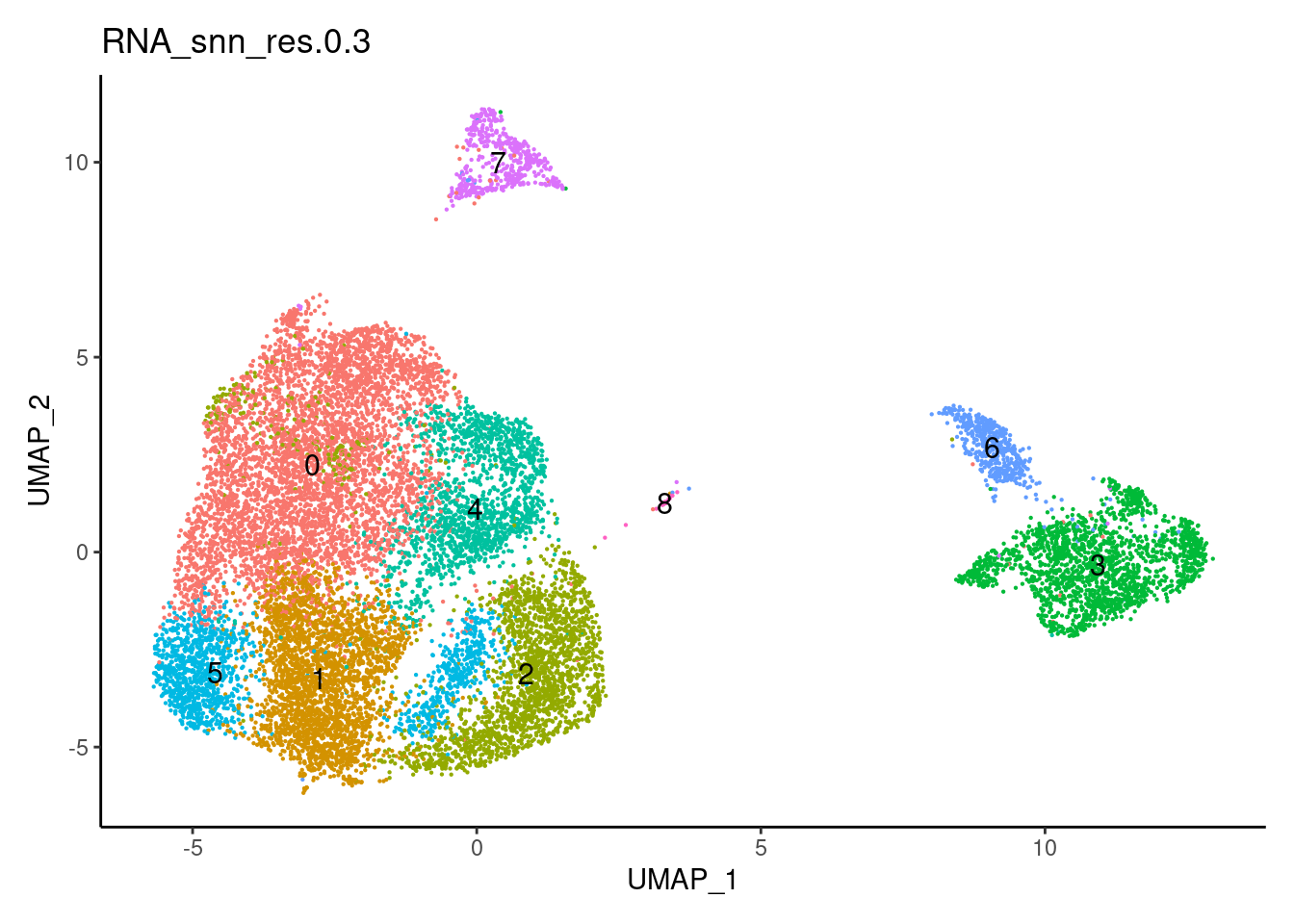
0.5
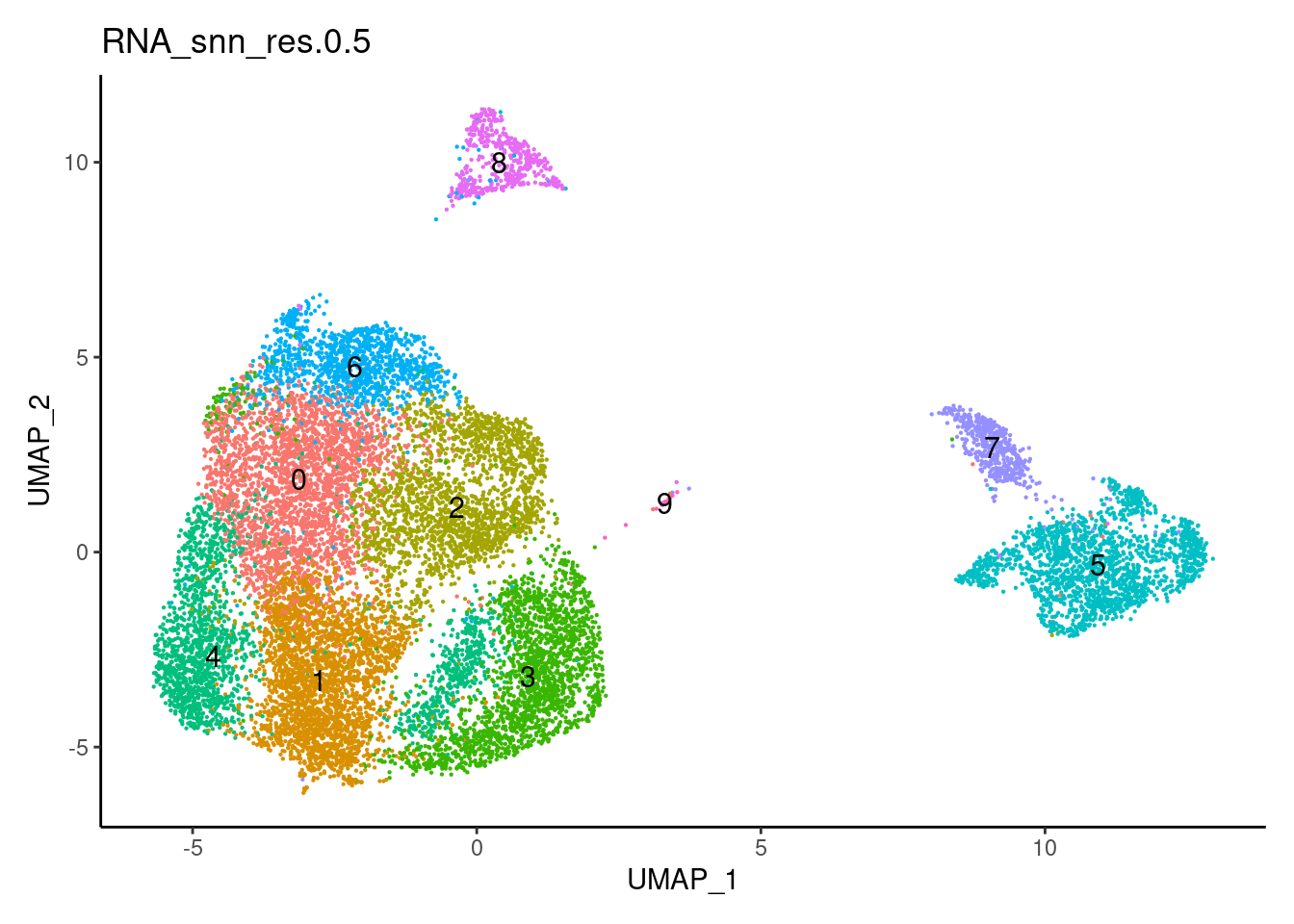
0.7
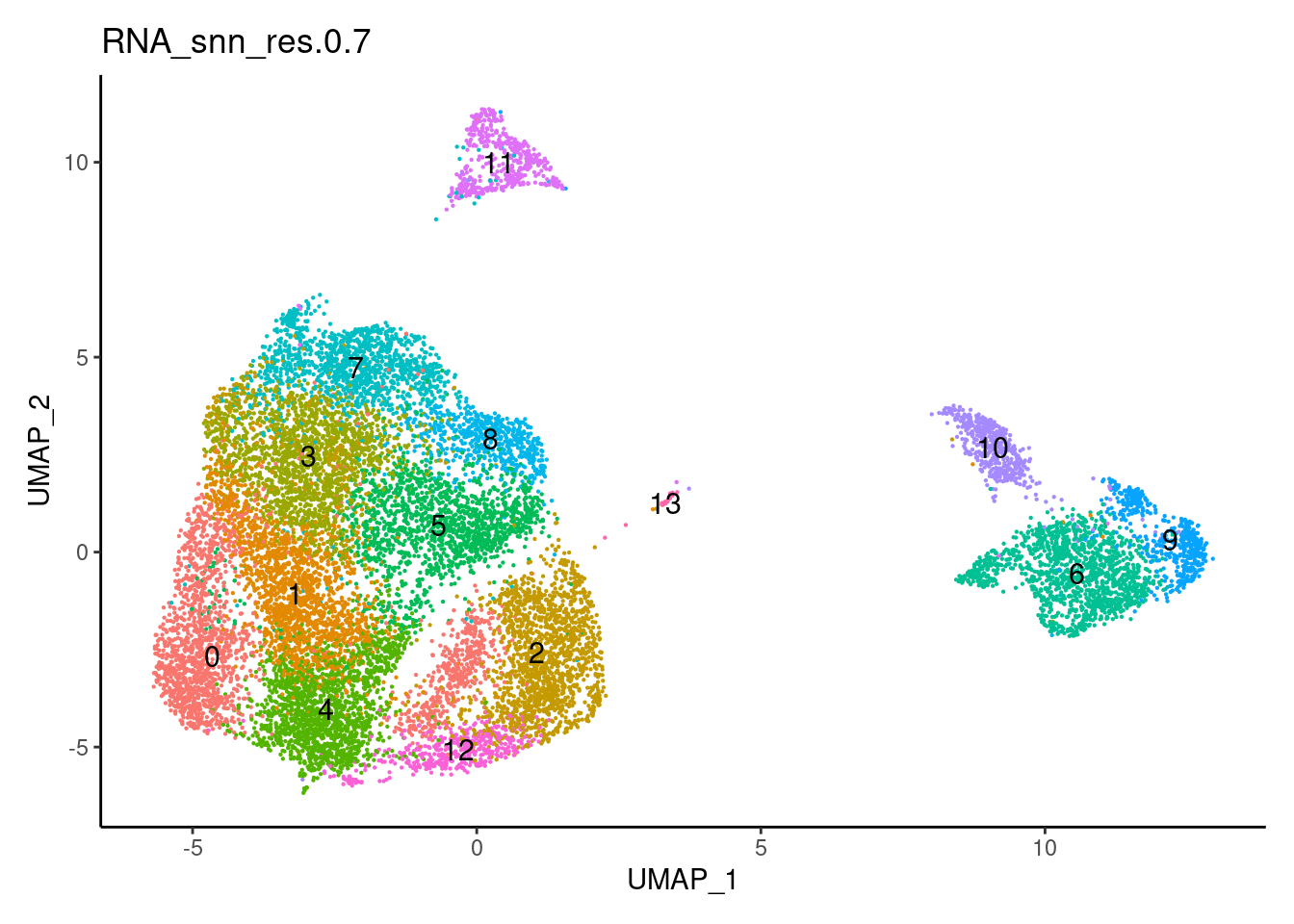
0.9
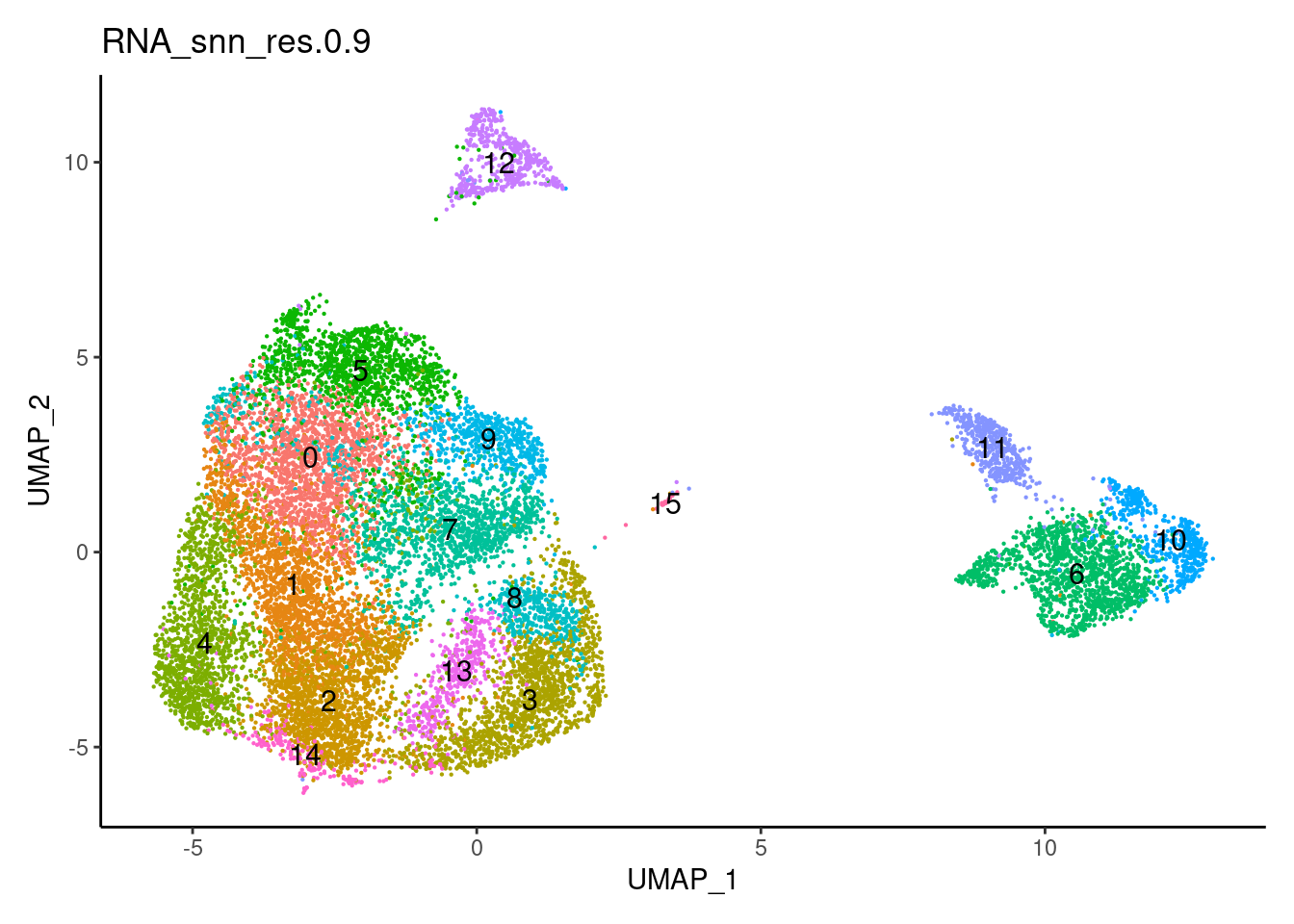
1.1
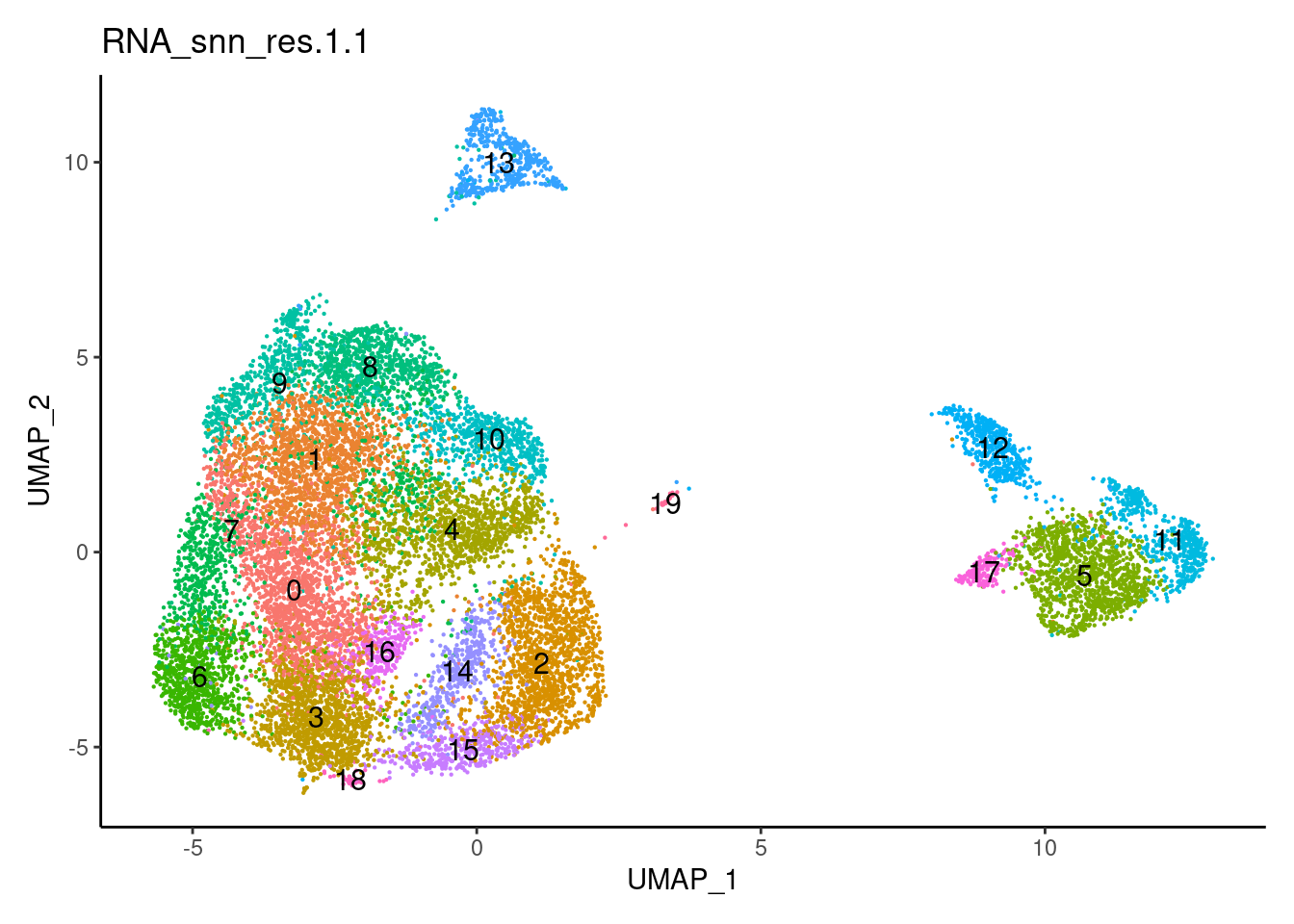
1.3
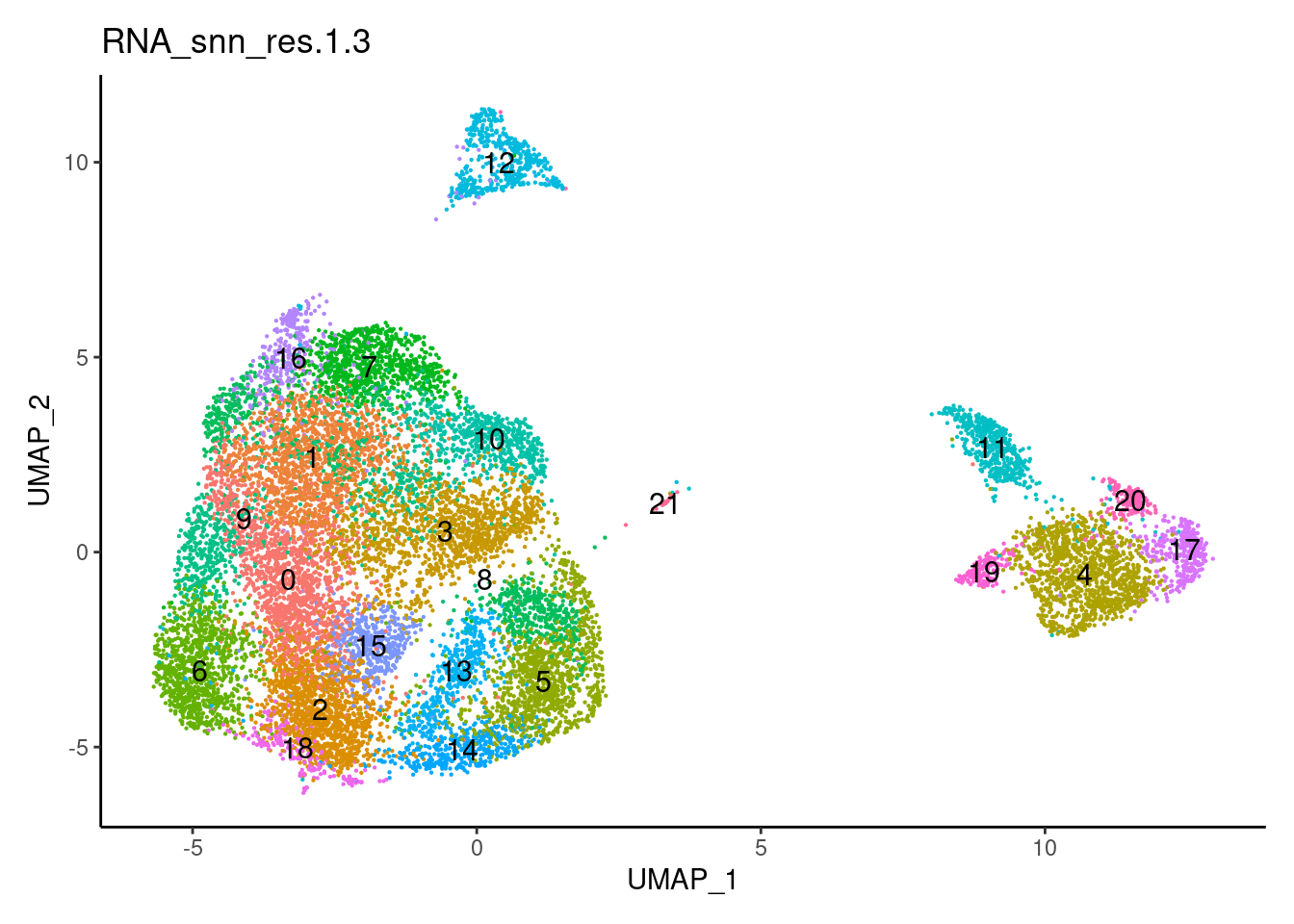
1.5
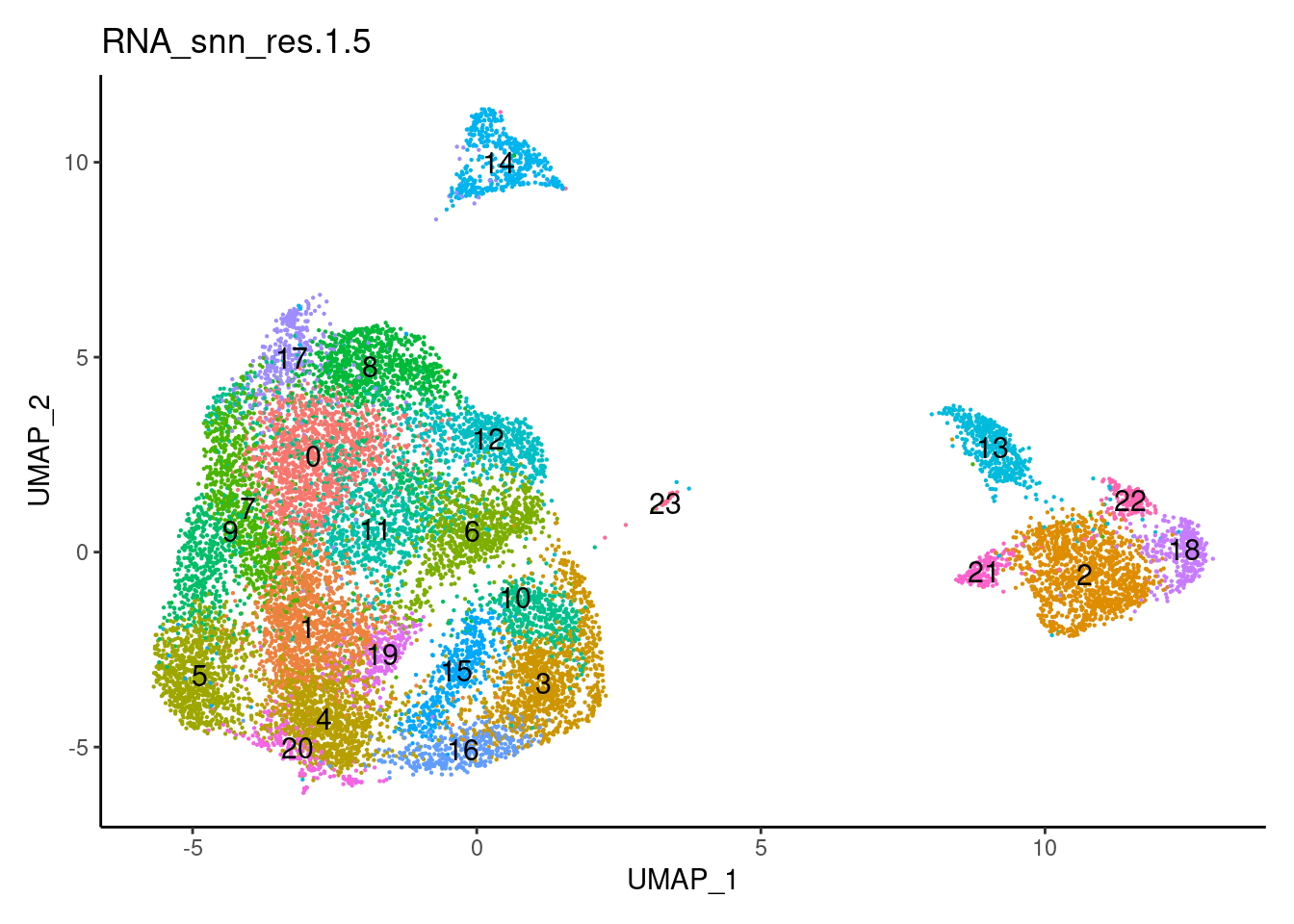
Clustree Resolutions
library(clustree)
clustree(plasmas, prefix = 'RNA_snn_res.') + guides(size = 'none', shape = 'none', edge_colour = FALSE, edge_alpha = FALSE) + theme(legend.position = 'right')## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.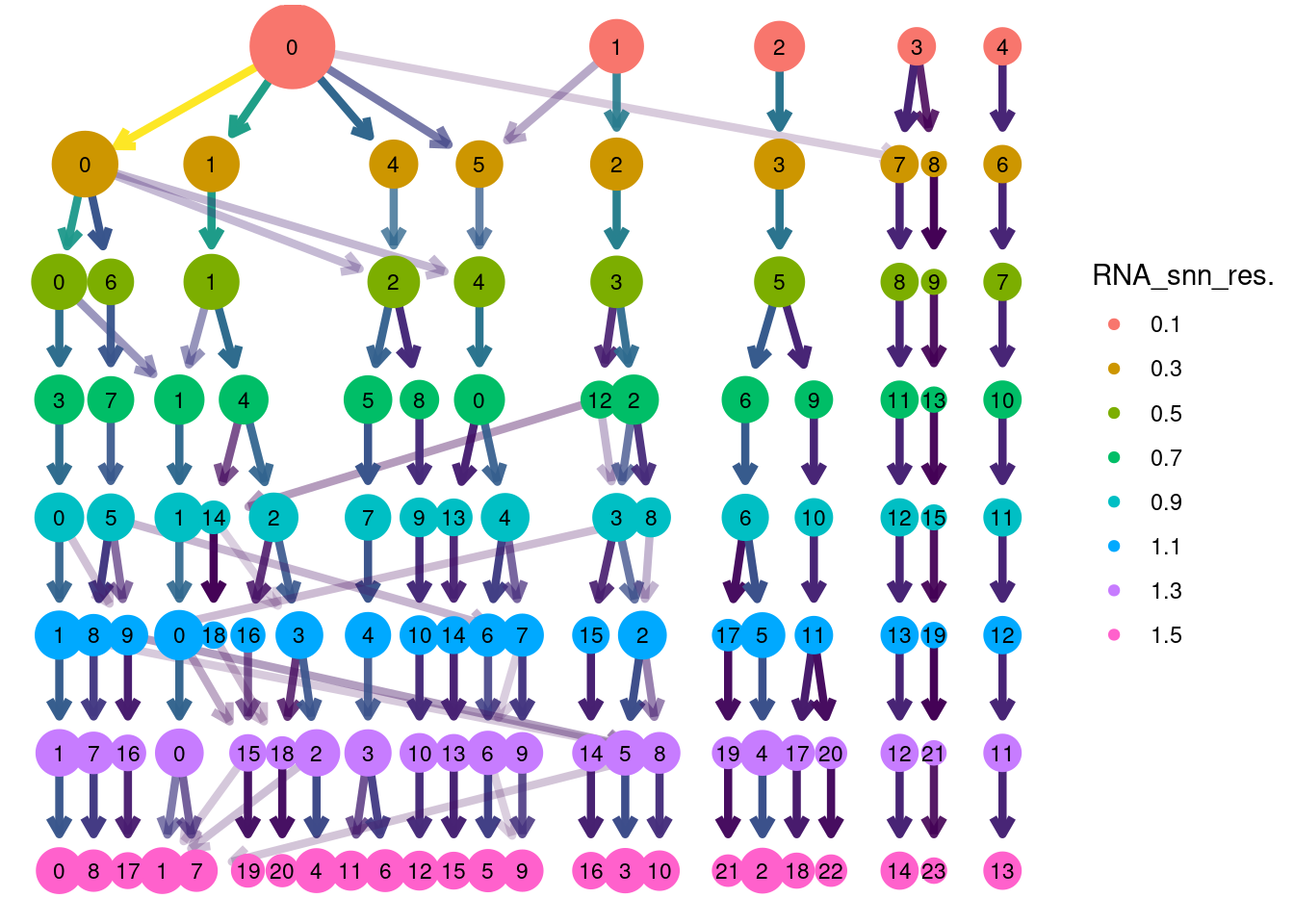
Top2 markers
setwd('~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/SUBSETS/')
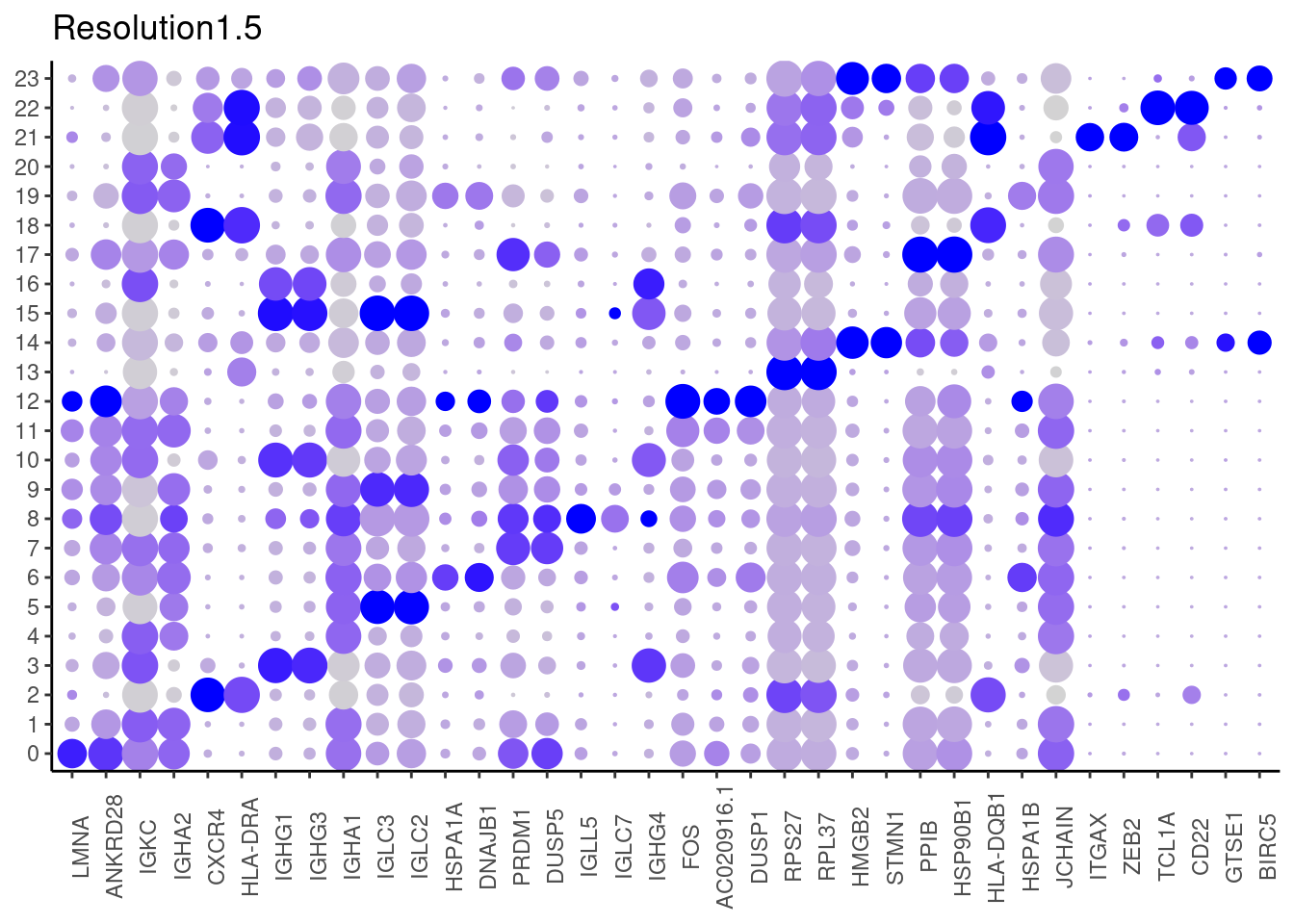
for(i in c('0.1','0.3','0.5','0.7','0.9','1.1','1.3','1.5')){
marker_genes <- read_delim(
paste0("Markers/BandPlasmacells/Markers_plasmas__markers_resolution_",i,".csv"),
delim = ";", escape_double = FALSE, locale = locale(decimal_mark = ",",
grouping_mark = "."), trim_ws = TRUE)
top2 <- marker_genes %>%
dplyr::group_by(cluster)%>%
dplyr::slice(1:2)
top2_g <- unique(top2$gene)
j <- DotPlot(plasmas, features = top2_g, group.by = paste0('RNA_snn_res.', i)) +
theme_classic() +
theme(axis.text.x = element_text(angle=90),
axis.title = element_blank()) +
NoLegend() +
labs(title = paste0('Resolution',i))
cat("##### ", i, "\n"); print(j); cat("\n\n")
}## Rows: 4290 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.0.1
## Rows: 5399 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.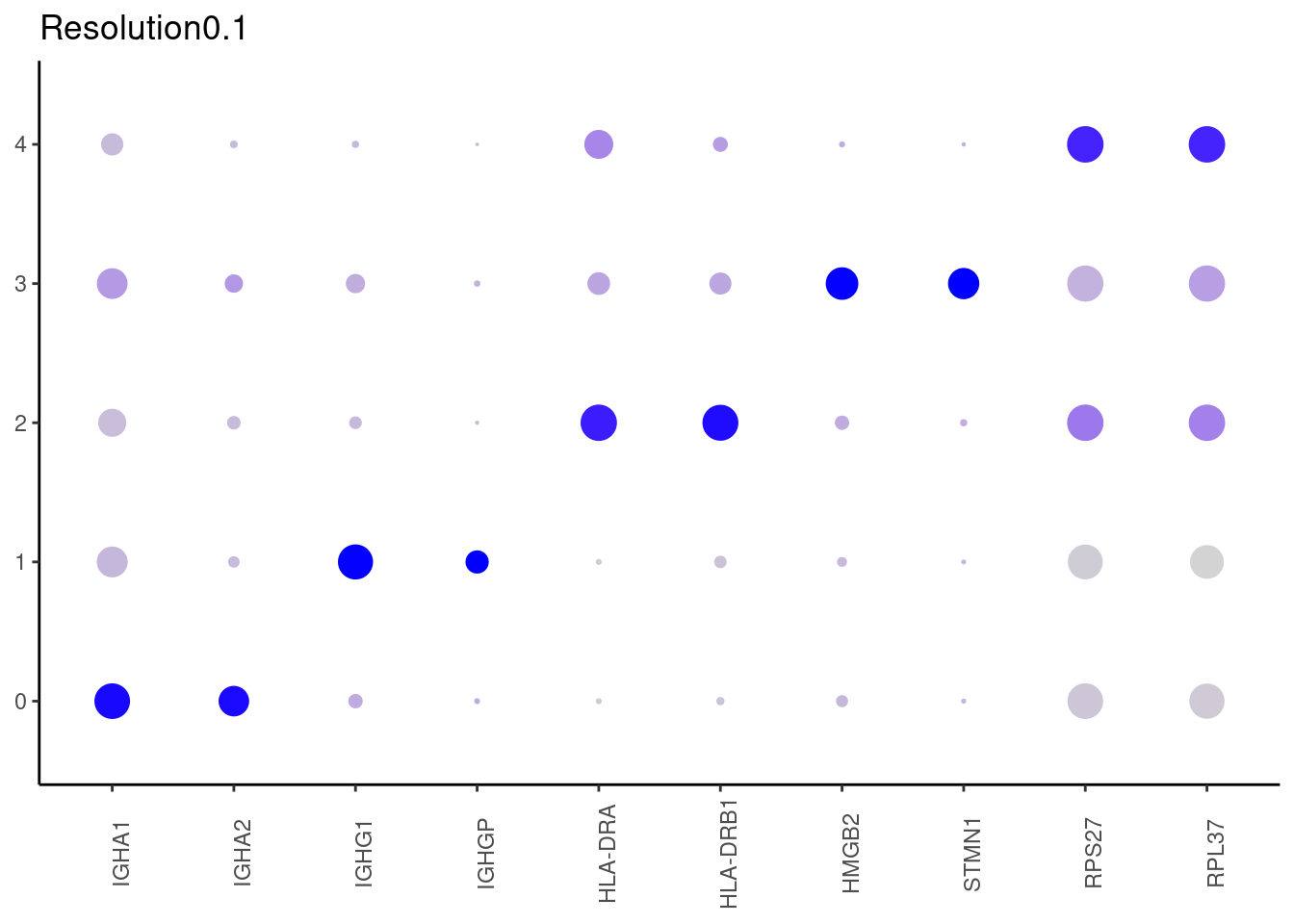 #####
0.3
#####
0.3
## Rows: 5985 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.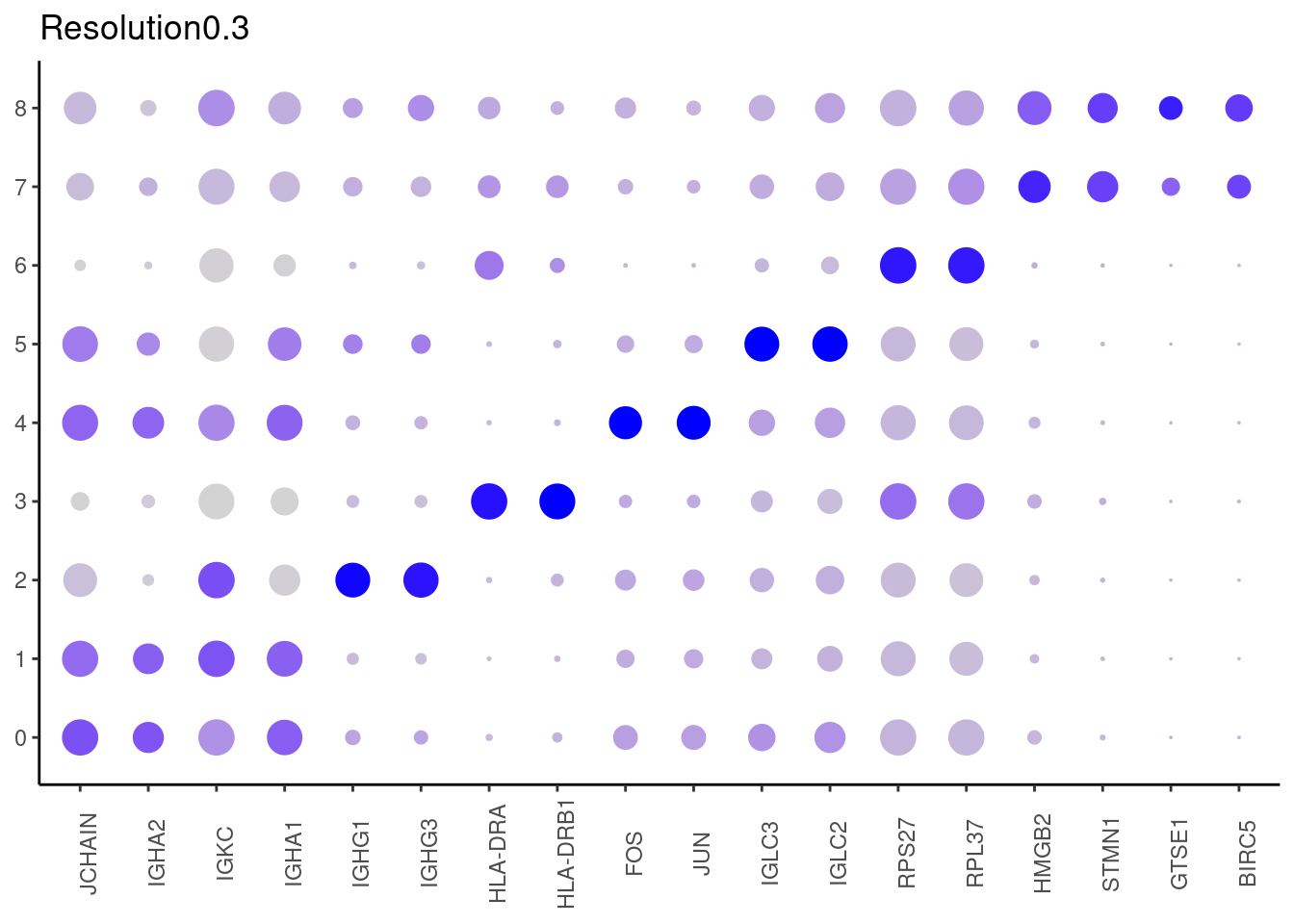 #####
0.5
#####
0.5
## Rows: 7352 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.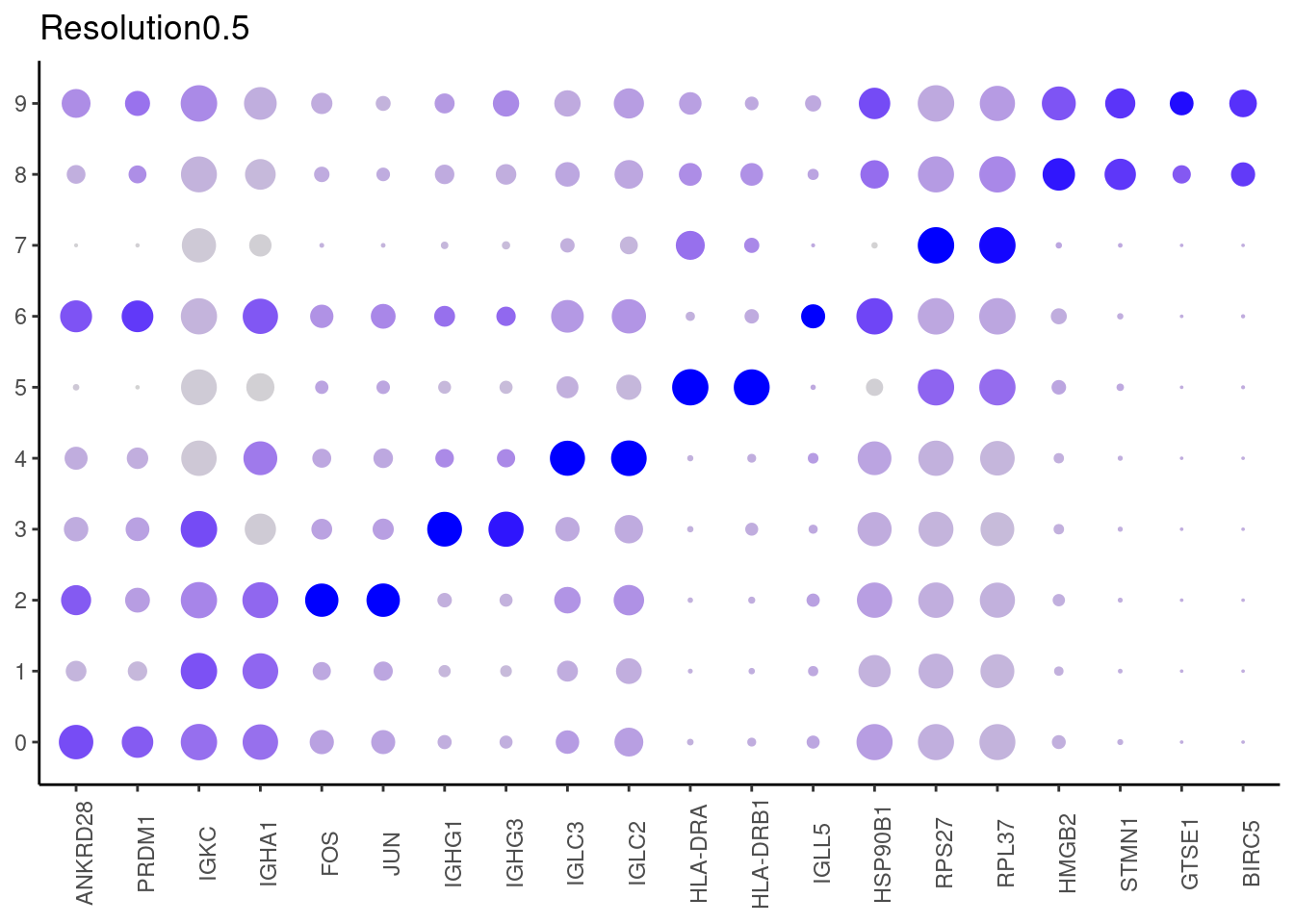 #####
0.7
#####
0.7
## Rows: 7297 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.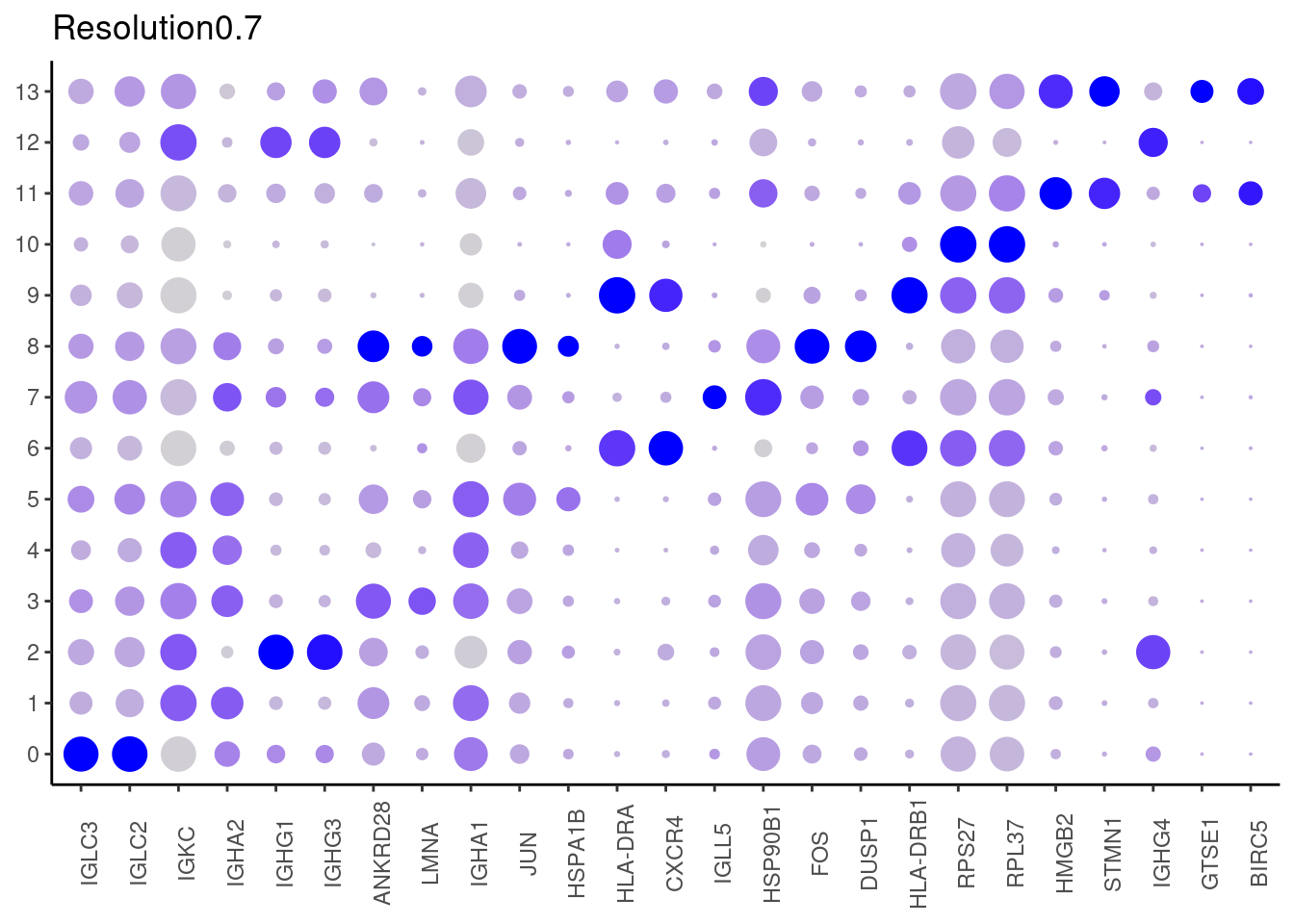 #####
0.9
#####
0.9
## Rows: 9789 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.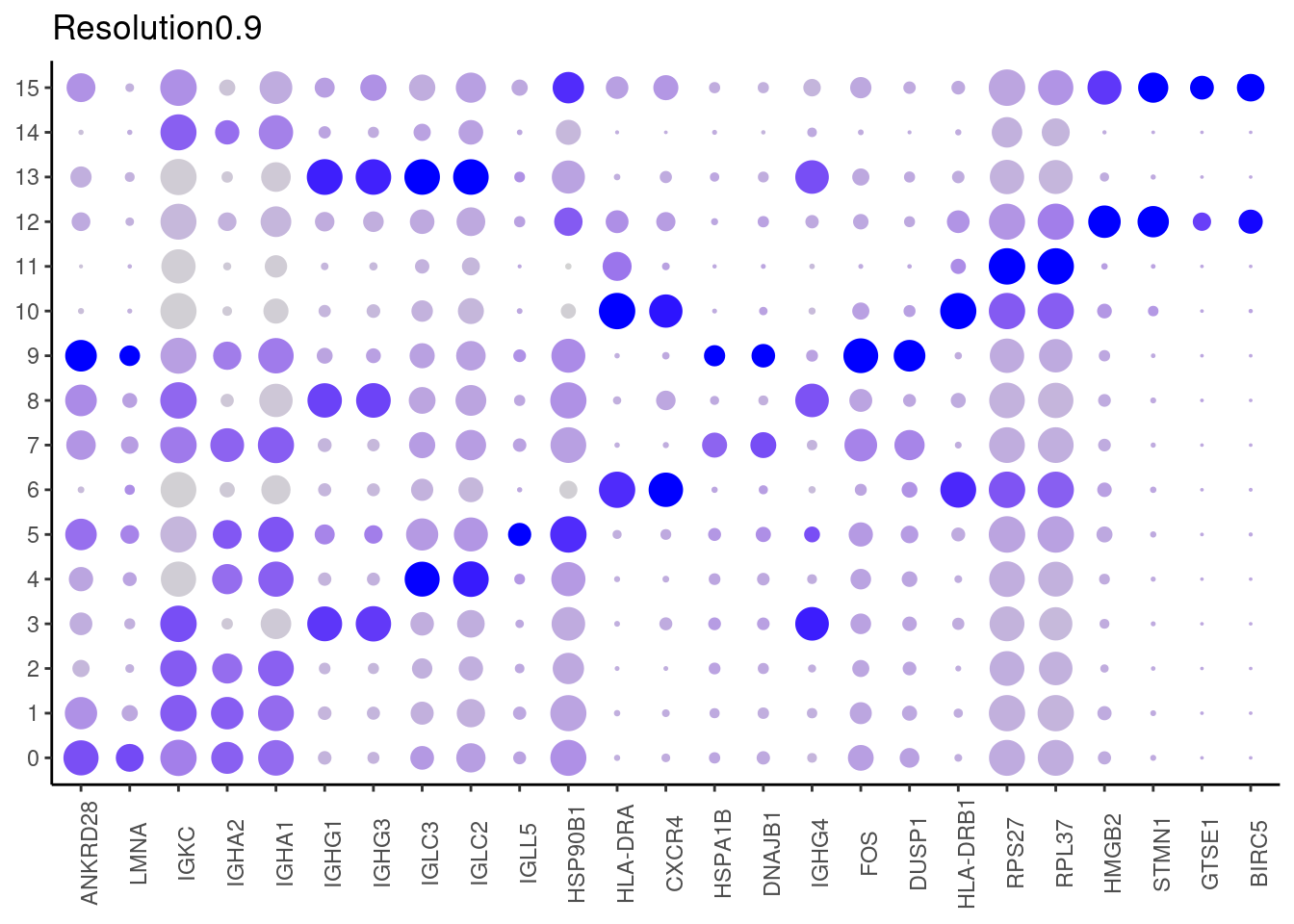 #####
1.1
#####
1.1
## Rows: 11267 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.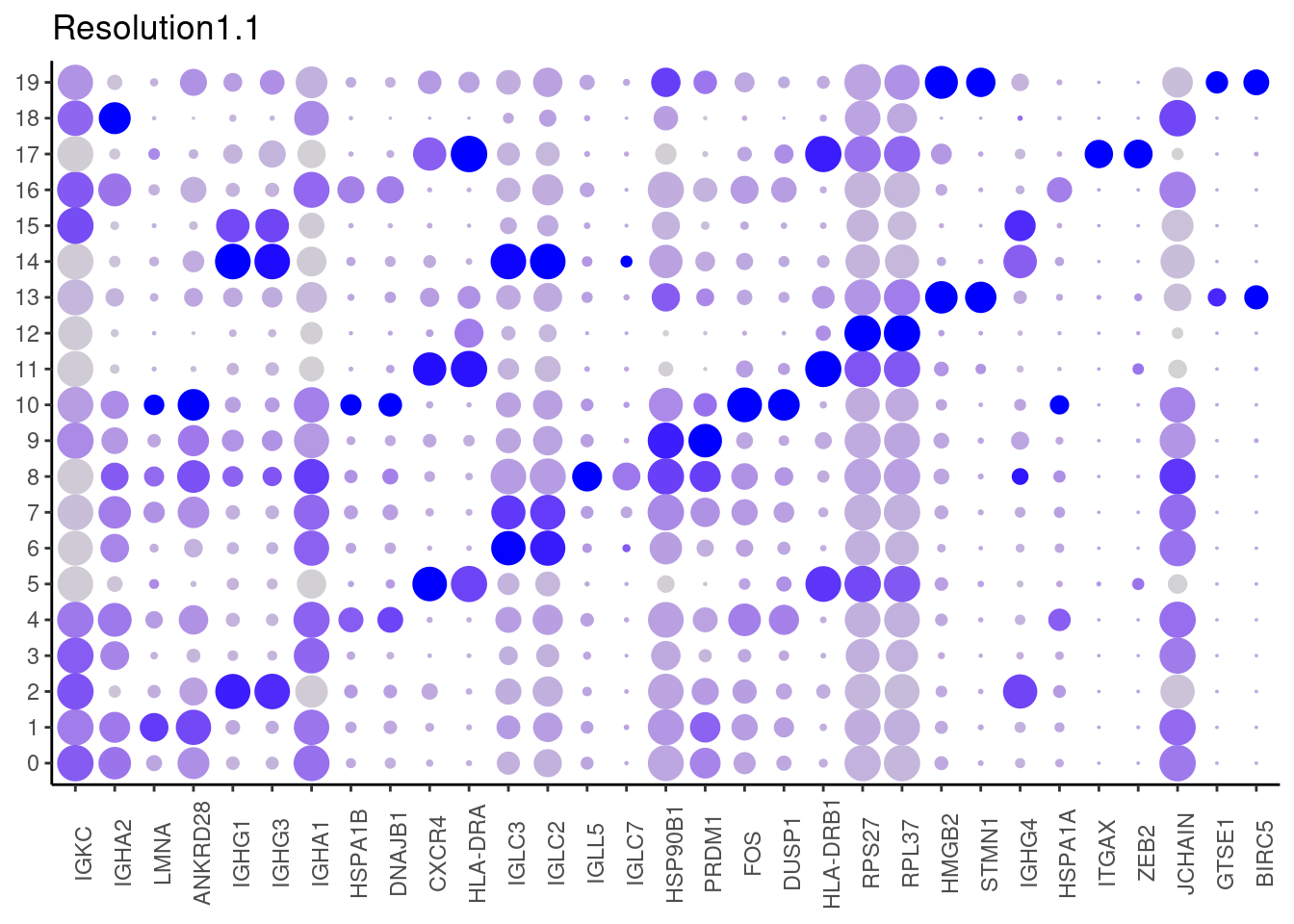 #####
1.3
#####
1.3
## Rows: 11440 Columns: 7
## ── Column specification ──────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## chr (1): gene
## dbl (6): p_val, avg_log2FC, pct.1, pct.2, p_val_adj, cluster
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.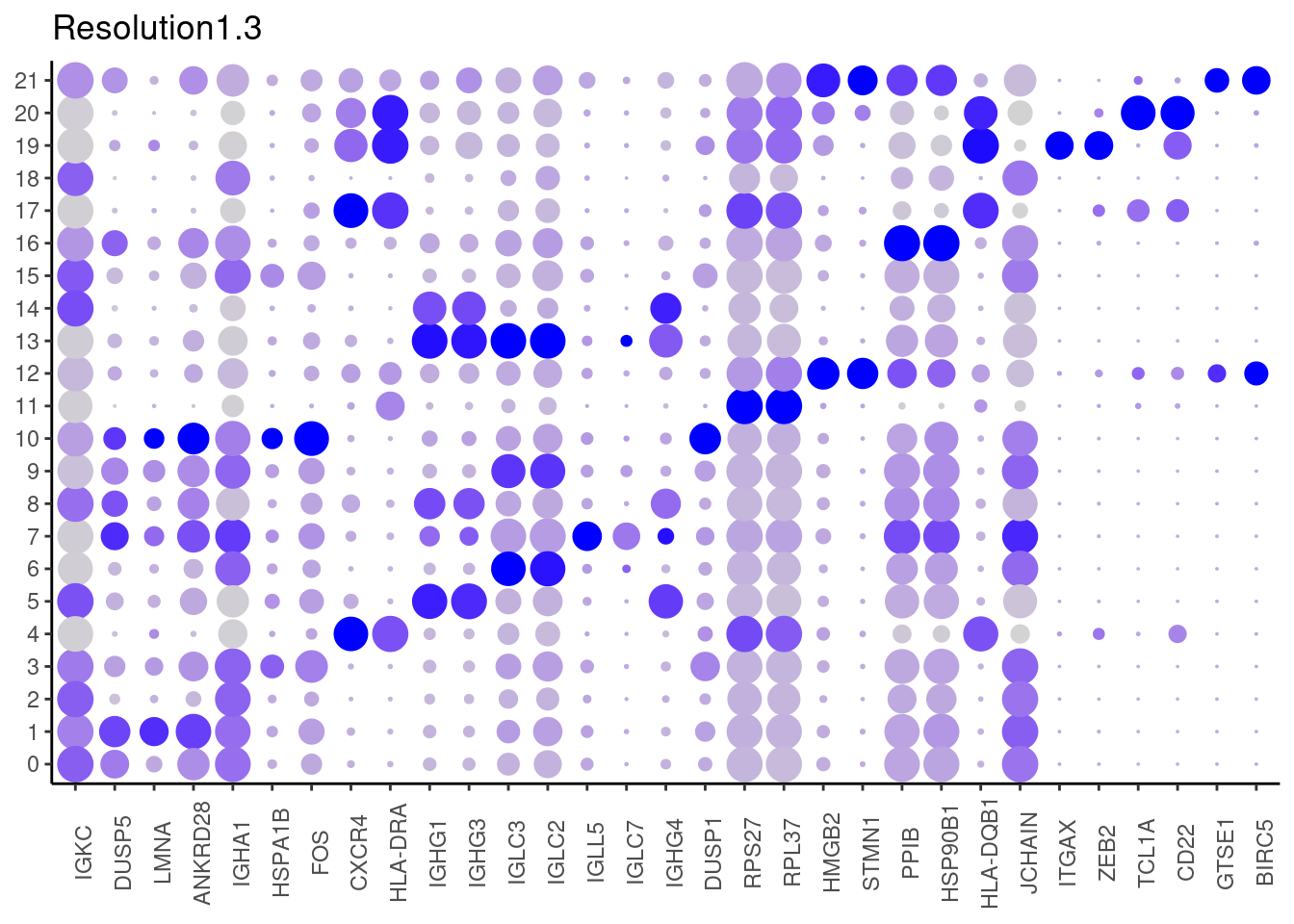 #####
1.5
#####
1.5
Annotation
plasmas$annotation_refined <- plyr::mapvalues(x = plasmas$RNA_snn_res.1.3,
from = 0:21,
to = c("PC IgA 1",
"PC IgA 2",
"PC IgA IgM",
"PC IgA heat shock 1",
"B cell",
"PC IgG 1",
"PC IgA Lambda 1",
"PC IGLL5",
"PC IgG 2",
"Plasmablast IgA Lambda 2",
"PC IER",
"B cell Ribhi",
"Cycling cells",
"Plasmablast IgG Lambda",
"Plasmablast IgG",
"PC IgA heat shock 2",
"PC IgA 3",
"Naïve B cell",
"PC IgA 4",
"Memory B cell",
"GC B cell",
"Cycling cells 2")
)
plasmas$annotation_intermediate <- plyr::mapvalues(x = plasmas$RNA_snn_res.1.3,
from = 0:21,
to = c("PC IgA",
"PC IgA",
"PC IgA",
"PC IgA heat shock",
"B cell",
"PC IgG",
"PC IgA",
"PC IgG",
"PC IgG",
"PC IgA",
"PC IER",
"B cell",
"Cycling cells",
"PC IgG",
"PC IgG",
"PC IgA heat shock",
"PC IgA",
"Naïve B cell",
"PC IgA",
"Memory B cell",
"GC B cell",
"Cycling cells"))
a <- DimPlot(plasmas, group.by = 'annotation_intermediate', label = T, repel=T, cols = cols_plasmas) + NoLegend()
b <- DimPlot(plasmas, group.by = 'annotation_refined', label = T, repel=T) + NoLegend()
a+b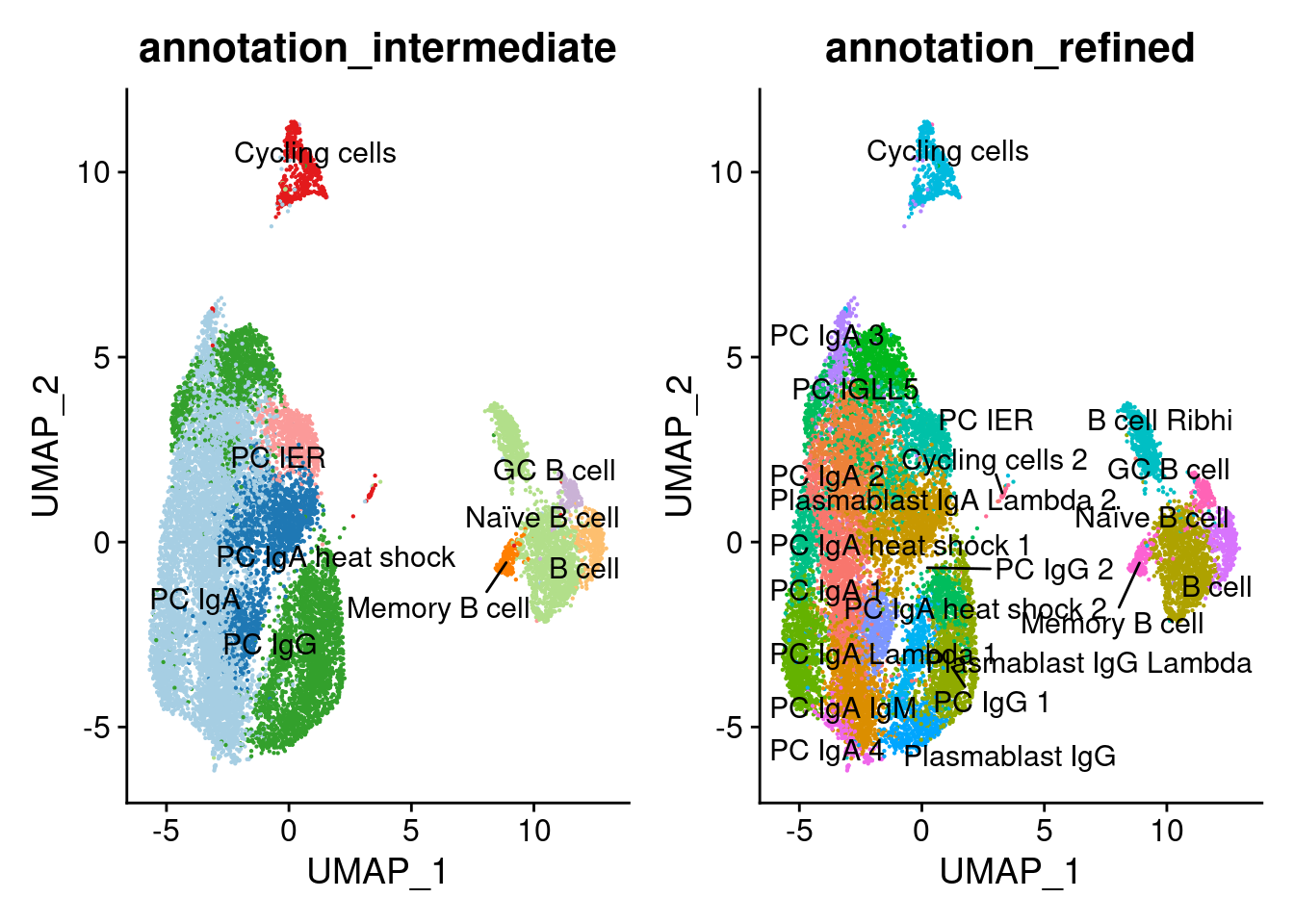
Save file
saveRDS(plasmas, file = '~/000_GitHub/ibd-bcn_single_cell/Analysis of our data/02_Samples_Together/SUBSETS/ON_THEIR_OWN/plasmas_annotated.RDS')
head(plasmas@meta.data)## orig.ident nCount_RNA nFeature_RNA sample doublet Health newname Health_2 percent.mt
## SC_002_AAACCTGAGGTCGGAT-1 SC 13513 1387 SC_002 singlet HC HC_SC_002 HC 1.628062
## SC_002_AAACCTGGTCGTCTTC-1 SC 2021 714 SC_002 singlet HC HC_SC_002 HC 10.836220
## SC_002_AAAGATGCACACCGAC-1 SC 6587 716 SC_002 singlet HC HC_SC_002 HC 2.429027
## SC_002_AAAGCAATCTGTGCAA-1 SC 16702 1298 SC_002 singlet HC HC_SC_002 HC 1.137588
## SC_002_AAATGCCTCAAGGCTT-1 SC 2249 662 SC_002 singlet HC HC_SC_002 HC 3.423744
## SC_002_AACTCAGAGCGTCTAT-1 SC 6172 906 SC_002 singlet HC HC_SC_002 HC 1.814647
## RNA_snn_res.0.1 seurat_clusters RNA_snn_res.0.3 RNA_snn_res.0.5 RNA_snn_res.0.7
## SC_002_AAACCTGAGGTCGGAT-1 0 12 4 2 8
## SC_002_AAACCTGGTCGTCTTC-1 2 2 3 5 6
## SC_002_AAAGATGCACACCGAC-1 0 4 1 1 4
## SC_002_AAAGCAATCTGTGCAA-1 0 6 4 2 5
## SC_002_AAATGCCTCAAGGCTT-1 4 13 6 7 10
## SC_002_AACTCAGAGCGTCTAT-1 0 12 4 2 8
## RNA_snn_res.0.9 RNA_snn_res.1.1 RNA_snn_res.1.3 RNA_snn_res.1.5 sample_name
## SC_002_AAACCTGAGGTCGGAT-1 9 10 10 12 HC 1
## SC_002_AAACCTGGTCGTCTTC-1 6 5 4 2 HC 1
## SC_002_AAAGATGCACACCGAC-1 2 3 2 4 HC 1
## SC_002_AAAGCAATCTGTGCAA-1 7 4 3 6 HC 1
## SC_002_AAATGCCTCAAGGCTT-1 11 12 11 13 HC 1
## SC_002_AACTCAGAGCGTCTAT-1 9 10 10 12 HC 1
## annotation_refined annotation_intermediate
## SC_002_AAACCTGAGGTCGGAT-1 PC IER PC IER
## SC_002_AAACCTGGTCGTCTTC-1 B cell B cell
## SC_002_AAAGATGCACACCGAC-1 PC IgA IgM PC IgA
## SC_002_AAAGCAATCTGTGCAA-1 PC IgA heat shock 1 PC IgA heat shock
## SC_002_AAATGCCTCAAGGCTT-1 B cell Ribhi B cell
## SC_002_AACTCAGAGCGTCTAT-1 PC IER PC IERsessionInfo()
sessionInfo()## R version 4.1.2 (2021-11-01)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.5 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
## LAPACK: /opt/R/4.1.2/lib/R/lib/libRlapack.so
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8 LC_COLLATE=C.UTF-8
## [5] LC_MONETARY=C.UTF-8 LC_MESSAGES=C LC_PAPER=C.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] Matrix_1.4-0 nnet_7.3-17 matchSCore2_0.1.0 harmony_0.1.0
## [5] Rcpp_1.0.9 rmarkdown_2.18 pandoc_0.1.0 readxl_1.3.1
## [9] magick_2.7.3 data.table_1.14.2 BiocParallel_1.28.3 RColorBrewer_1.1-3
## [13] ggrepel_0.9.1 ggrastr_1.0.1 usethis_2.1.5 clustree_0.4.4
## [17] ggraph_2.0.5 readr_2.1.2 dplyr_1.0.10 cowplot_1.1.1
## [21] reshape_0.8.8 formulaic_0.0.8 patchwork_1.1.2 MASS_7.3-55
## [25] viridis_0.6.2 viridisLite_0.4.1 scDblFinder_1.8.0 scran_1.22.1
## [29] scater_1.22.0 scuttle_1.4.0 celda_1.10.0 beepr_1.3
## [33] DropletUtils_1.14.2 SingleCellExperiment_1.16.0 SummarizedExperiment_1.24.0 Biobase_2.54.0
## [37] GenomicRanges_1.46.1 GenomeInfoDb_1.30.1 IRanges_2.28.0 S4Vectors_0.32.4
## [41] BiocGenerics_0.40.0 MatrixGenerics_1.6.0 matrixStats_0.62.0 ggplot2_3.3.6
## [45] plyr_1.8.7 sp_1.5-0 SeuratObject_4.1.1 Seurat_4.1.0.9007
##
## loaded via a namespace (and not attached):
## [1] rsvd_1.0.5 ica_1.0-3 corpcor_1.6.10 assertive.properties_0.0-4
## [5] foreach_1.5.2 lmtest_0.9-40 rprojroot_2.0.3 crayon_1.5.2
## [9] spatstat.core_2.4-4 rhdf5filters_1.6.0 backports_1.4.1 nlme_3.1-155
## [13] rlang_1.0.6 XVector_0.34.0 ROCR_1.0-11 irlba_2.3.5
## [17] SparseM_1.81 limma_3.50.3 xgboost_1.5.0.2 rjson_0.2.21
## [21] bit64_4.0.5 glue_1.6.2 sctransform_0.3.4 parallel_4.1.2
## [25] vipor_0.4.5 spatstat.sparse_2.1-1 AnnotationDbi_1.56.2 spatstat.geom_2.4-0
## [29] tidyselect_1.1.2 fitdistrplus_1.1-8 tidyr_1.2.1 assertive.types_0.0-3
## [33] zoo_1.8-10 org.Mm.eg.db_3.14.0 xtable_1.8-4 magrittr_2.0.3
## [37] evaluate_0.18 cli_3.4.0 zlibbioc_1.40.0 rstudioapi_0.13
## [41] miniUI_0.1.1.1 bslib_0.4.1 rpart_4.1.16 RcppEigen_0.3.3.9.2
## [45] shiny_1.7.3 BiocSingular_1.10.0 xfun_0.34 clue_0.3-60
## [49] cluster_2.1.2 tidygraph_1.2.1 KEGGREST_1.34.0 tibble_3.1.8
## [53] listenv_0.8.0 Biostrings_2.62.0 png_0.1-7 future_1.28.0
## [57] withr_2.5.0 bitops_1.0-7 ggforce_0.3.3 cellranger_1.1.0
## [61] assertive.base_0.0-9 dqrng_0.3.0 pillar_1.8.1 GlobalOptions_0.1.2
## [65] cachem_1.0.6 fs_1.5.2 GetoptLong_1.0.5 DelayedMatrixStats_1.16.0
## [69] vctrs_0.4.1 ellipsis_0.3.2 generics_0.1.3 tools_4.1.2
## [73] beeswarm_0.4.0 munsell_0.5.0 tweenr_1.0.2 DelayedArray_0.20.0
## [77] fastmap_1.1.0 compiler_4.1.2 pkgload_1.2.4 abind_1.4-5
## [81] httpuv_1.6.6 plotly_4.10.0 rgeos_0.5-9 GenomeInfoDbData_1.2.7
## [85] gridExtra_2.3 enrichR_3.0 edgeR_3.36.0 lattice_0.20-45
## [89] deldir_1.0-6 utf8_1.2.2 later_1.3.0 jsonlite_1.8.3
## [93] multipanelfigure_2.1.2 scales_1.2.1 graph_1.72.0 ScaledMatrix_1.2.0
## [97] pbapply_1.5-0 sparseMatrixStats_1.6.0 lazyeval_0.2.2 promises_1.2.0.1
## [101] doParallel_1.0.17 R.utils_2.12.0 goftest_1.2-3 checkmate_2.0.0
## [105] spatstat.utils_2.3-1 reticulate_1.26 textshaping_0.3.6 statmod_1.4.36
## [109] Rtsne_0.16 uwot_0.1.14 igraph_1.3.4 HDF5Array_1.22.1
## [113] survival_3.2-13 rsconnect_0.8.25 yaml_2.3.6 systemfonts_1.0.4
## [117] htmltools_0.5.3 memoise_2.0.1 locfit_1.5-9.6 graphlayouts_0.8.0
## [121] digest_0.6.30 assertthat_0.2.1 rappdirs_0.3.3 mime_0.12
## [125] RSQLite_2.2.17 future.apply_1.9.1 blob_1.2.3 R.oo_1.25.0
## [129] ragg_1.2.1 splines_4.1.2 labeling_0.4.2 Rhdf5lib_1.16.0
## [133] RCurl_1.98-1.8 assertive.numbers_0.0-2 hms_1.1.1 rhdf5_2.38.1
## [137] colorspace_2.0-3 ggbeeswarm_0.6.0 shape_1.4.6 assertive.files_0.0-2
## [141] sass_0.4.2 RANN_2.6.1 circlize_0.4.14 audio_0.1-10
## [145] fansi_1.0.3 tzdb_0.2.0 brio_1.1.3 parallelly_1.32.1
## [149] R6_2.5.1 grid_4.1.2 ggridges_0.5.3 lifecycle_1.0.3
## [153] formatR_1.12 bluster_1.4.0 curl_4.3.2 jquerylib_0.1.4
## [157] leiden_0.4.3 testthat_3.1.2 desc_1.4.0 RcppAnnoy_0.0.19
## [161] org.Hs.eg.db_3.14.0 iterators_1.0.14 stringr_1.4.1 topGO_2.46.0
## [165] htmlwidgets_1.5.4 beachmat_2.10.0 polyclip_1.10-0 purrr_0.3.4
## [169] gridGraphics_0.5-1 ComplexHeatmap_2.10.0 mgcv_1.8-38 globals_0.16.1
## [173] spatstat.random_2.2-0 progressr_0.11.0 codetools_0.2-18 GO.db_3.14.0
## [177] metapod_1.2.0 MCMCprecision_0.4.0 R.methodsS3_1.8.2 gtable_0.3.1
## [181] DBI_1.1.3 highr_0.9 tensor_1.5 httr_1.4.4
## [185] KernSmooth_2.23-20 vroom_1.5.7 stringi_1.7.8 reshape2_1.4.4
## [189] farver_2.1.1 combinat_0.0-8 BiocNeighbors_1.12.0 scattermore_0.8
## [193] bit_4.0.4 spatstat.data_2.2-0 pkgconfig_2.0.3 corrplot_0.92
## [197] knitr_1.40